
Blood Transcriptome Analysis of Beef Cow with Different Parity Revealed Candidate Genes and Gene Networks Regulating the Postpartum Diseases

 , ,
, ,
Abstract
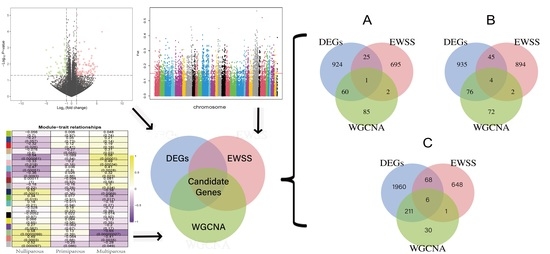
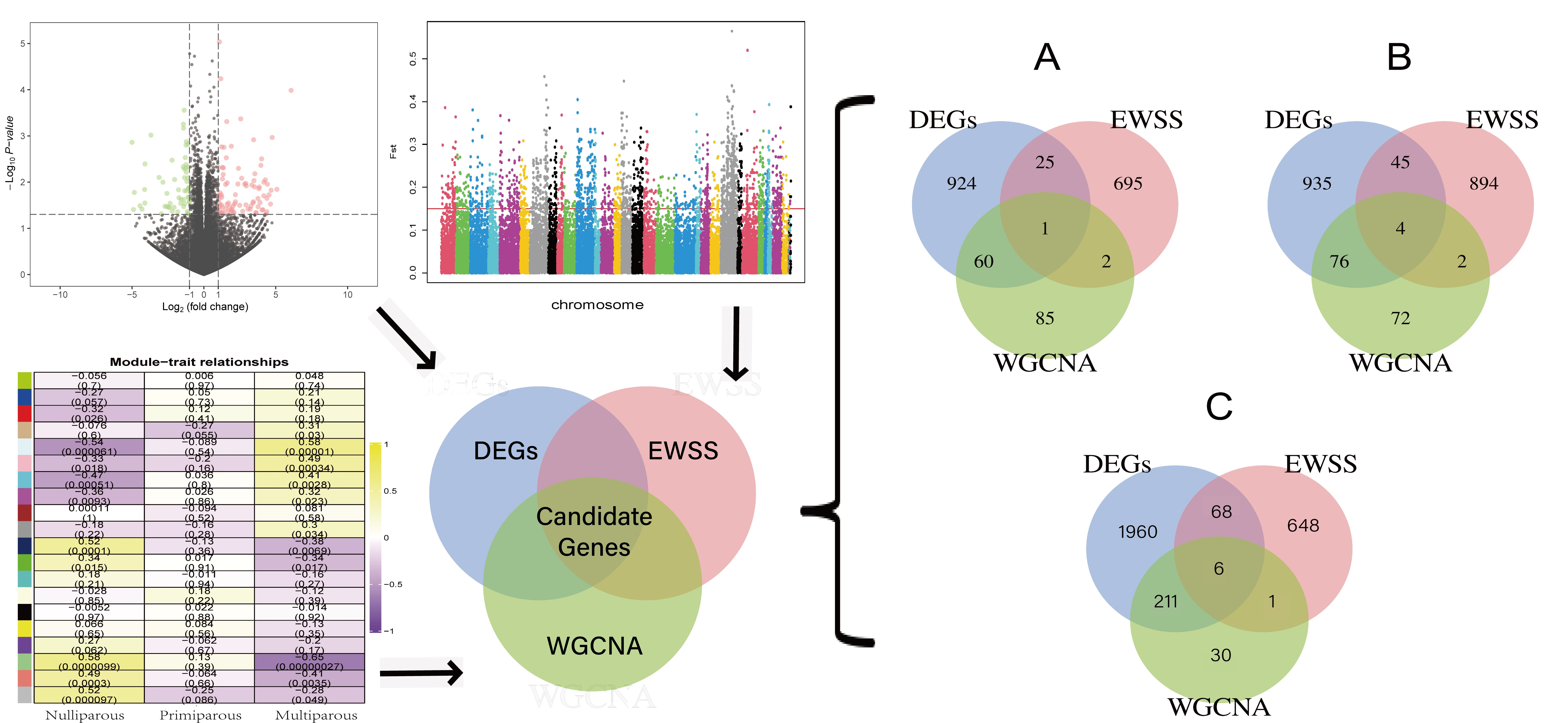
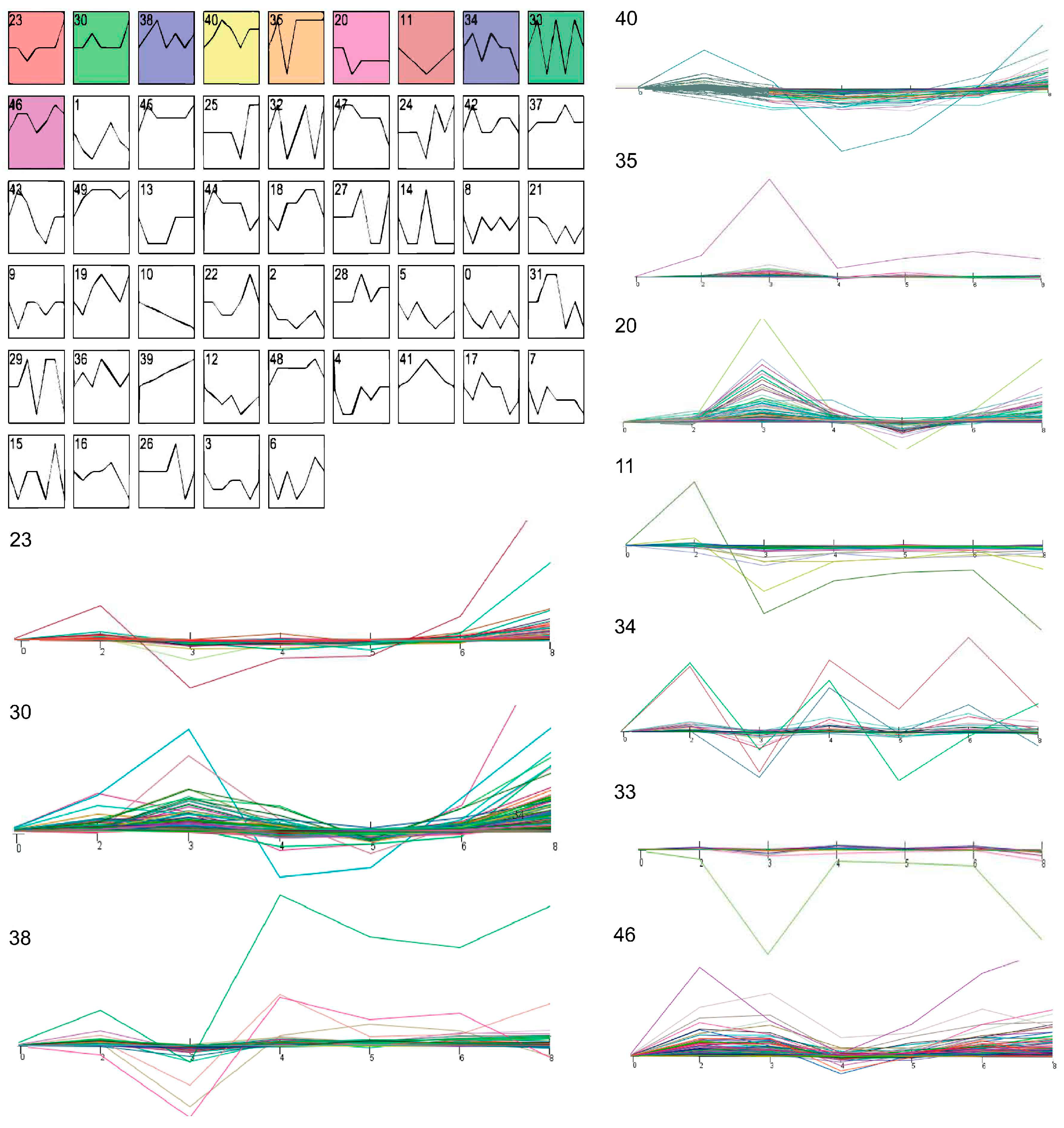
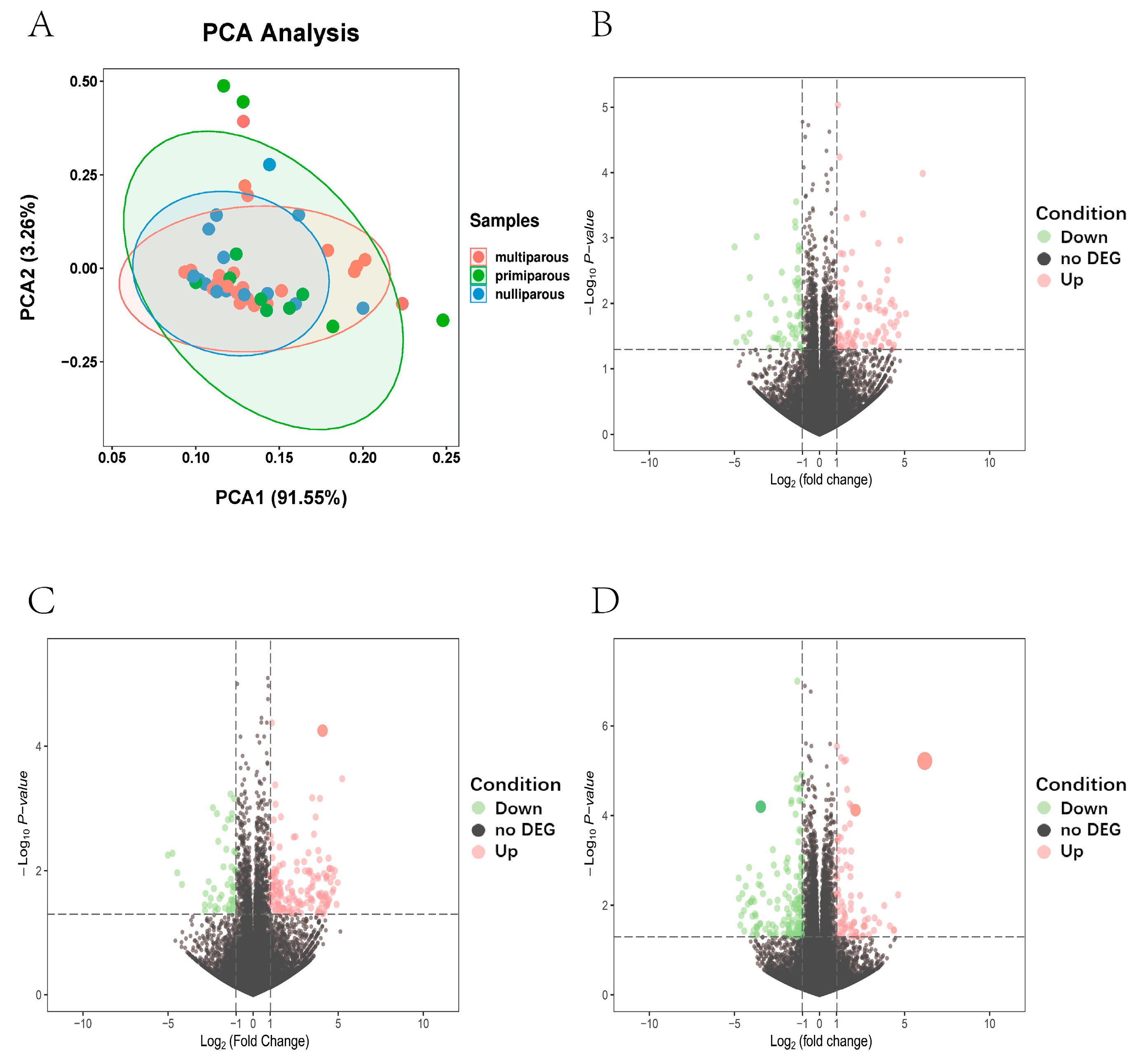
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Laboratory Animals and Feeding Management
2.2. Sequencing Data Analysis
2.3. Short Time-Series Expression Miner Analysis
2.4. Differentially Expressed Genes Analysis
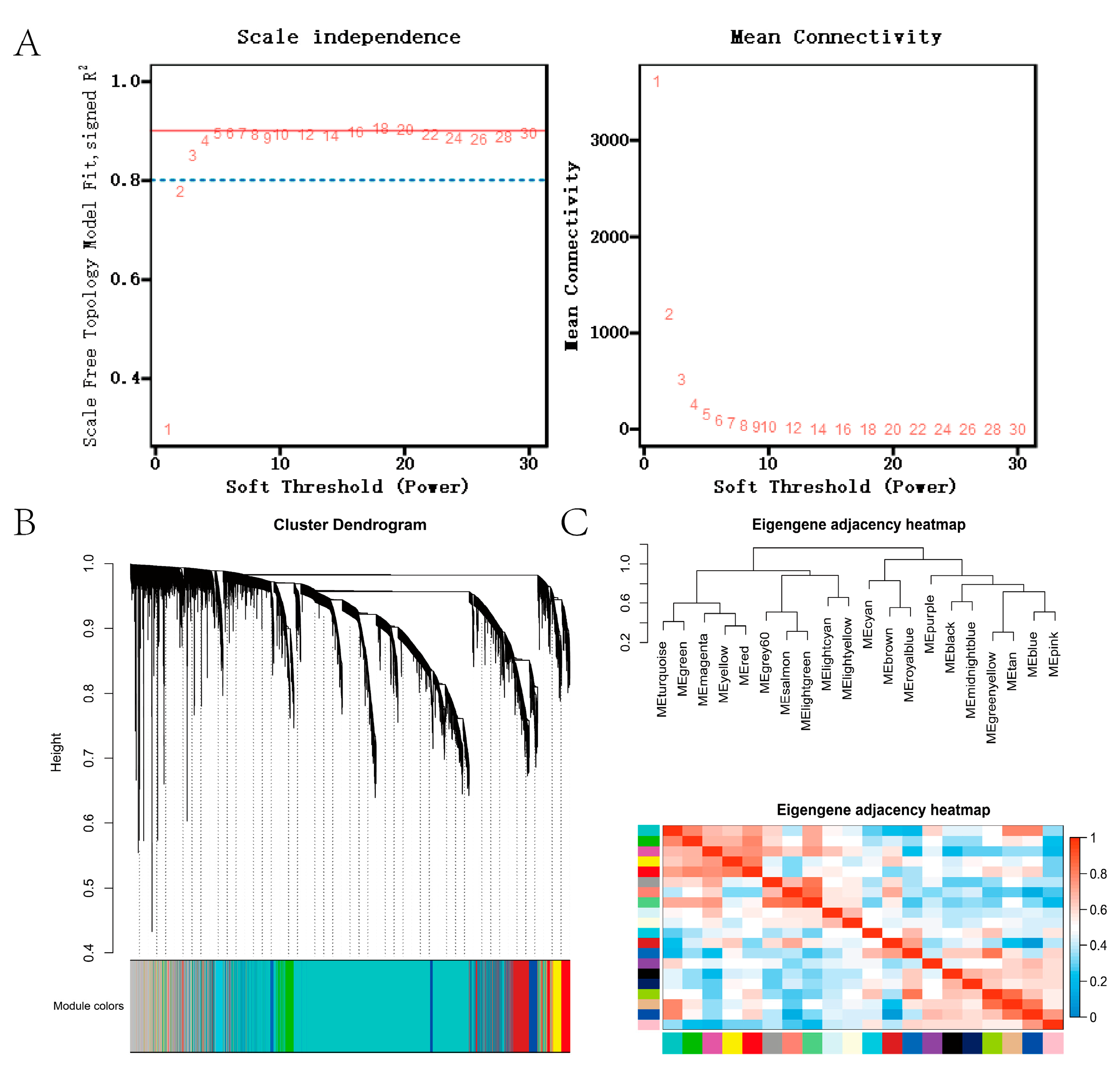
2.5. Co-Expression Network Analysis
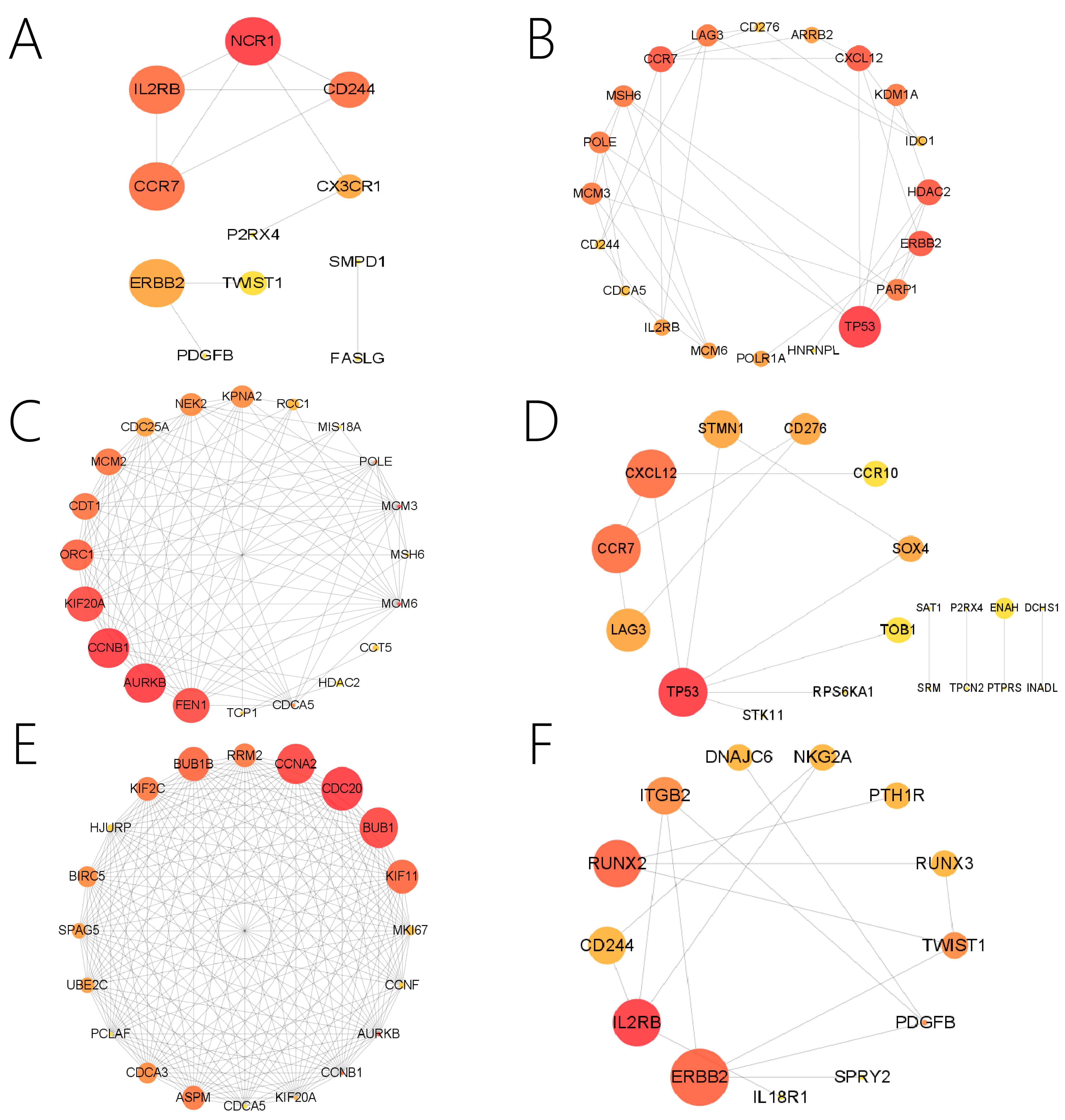
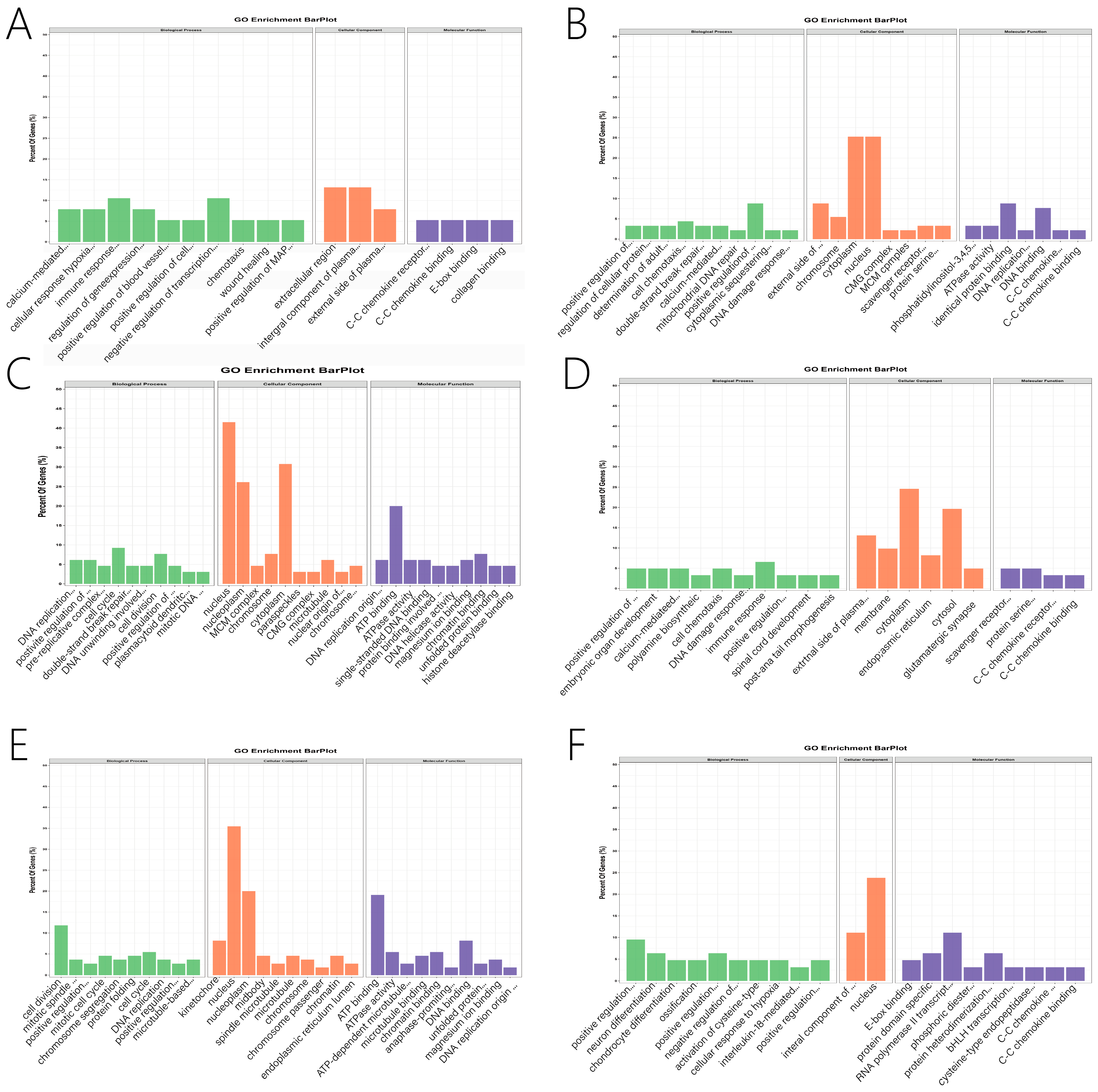
2.6. Function Enrichment and PPI Analysis
2.7. Exon Wide Selection Signature
3. Results
3.1. Data Analysis of Transcriptome
3.2. Differentially Expressed Genes across Three Periods
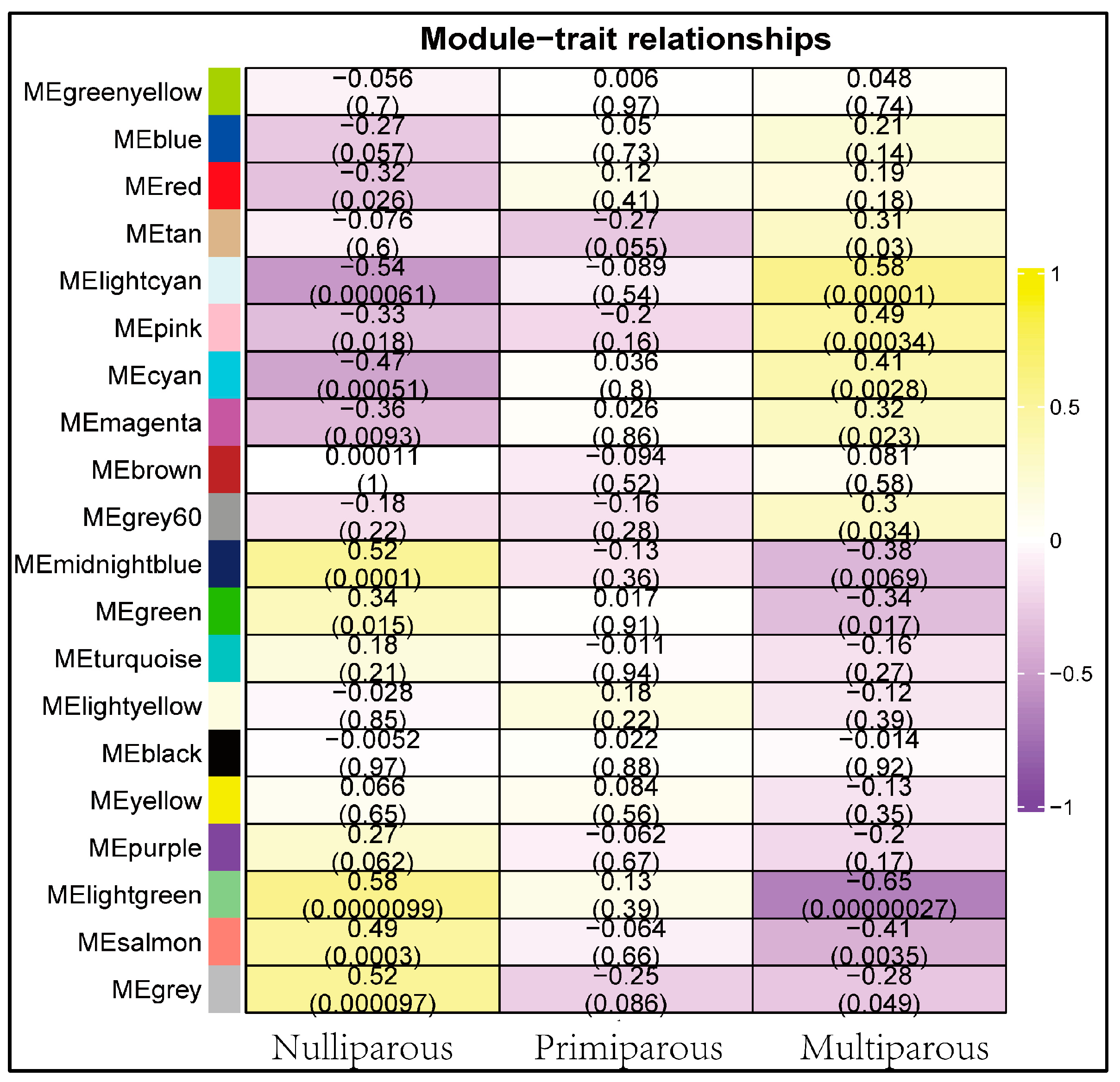
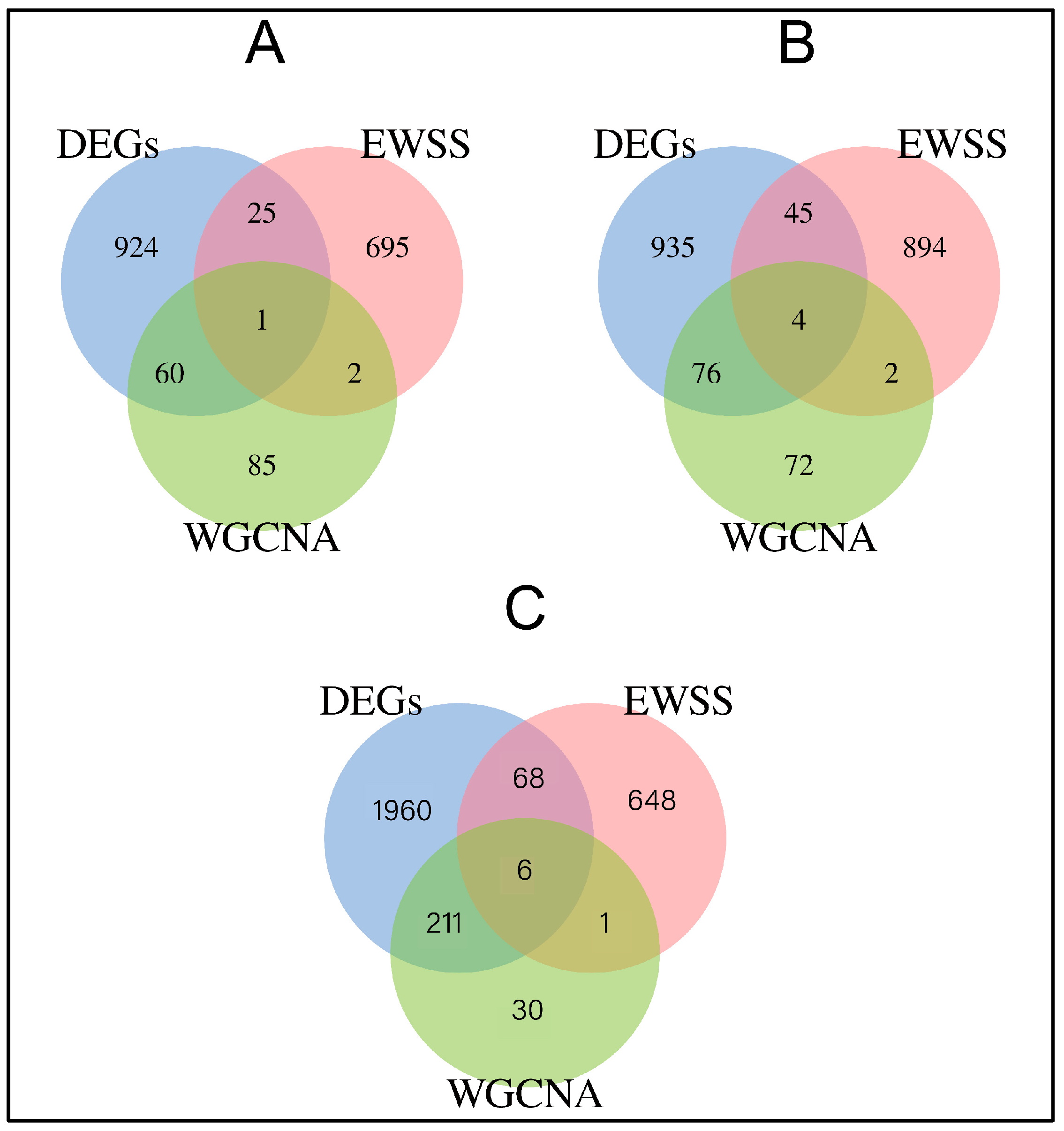
3.3. Co-Expression Network Construction and Module Detection
3.4. Period-Specific Module Identification
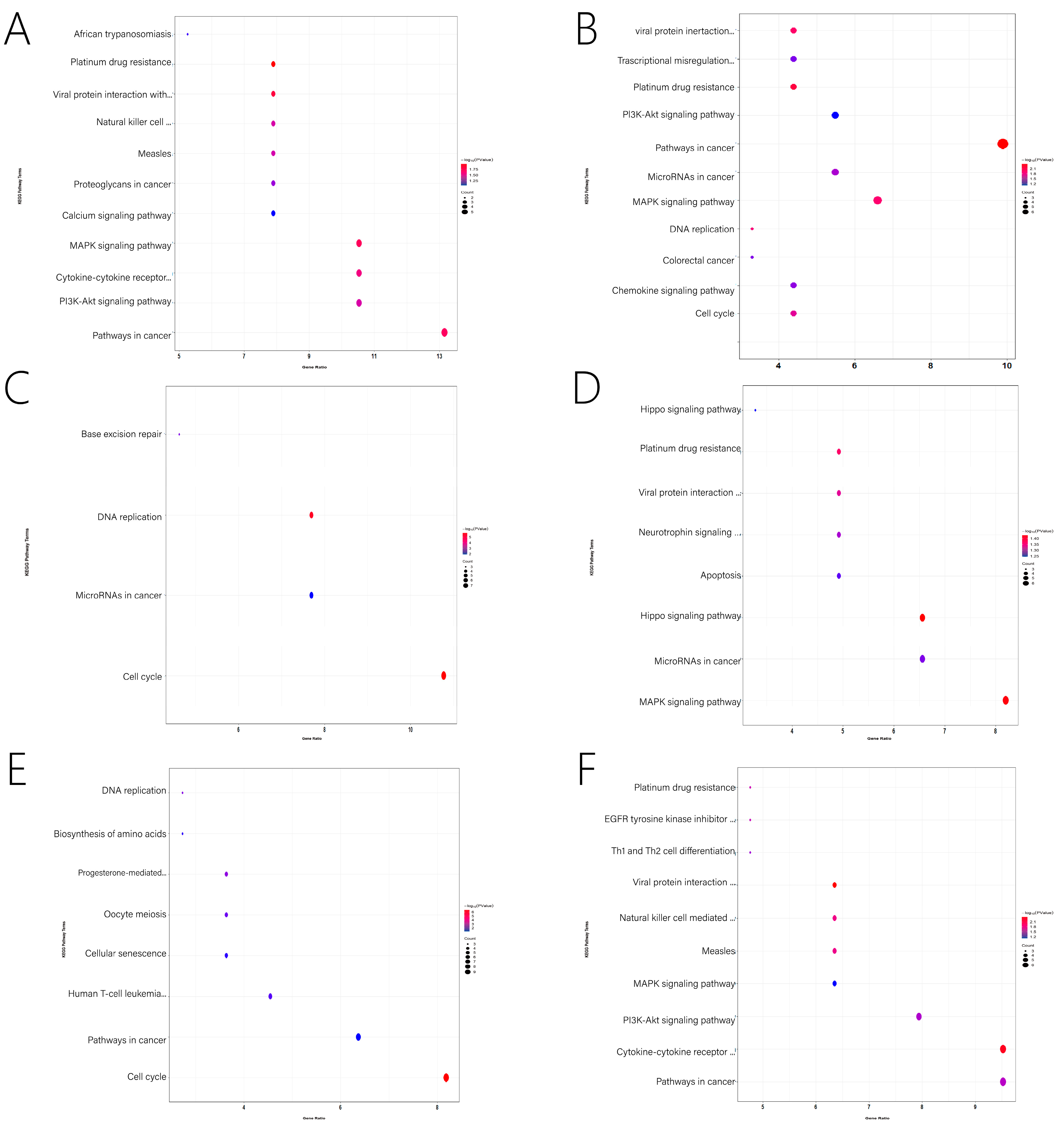
3.5. Function Enrichment Analysis
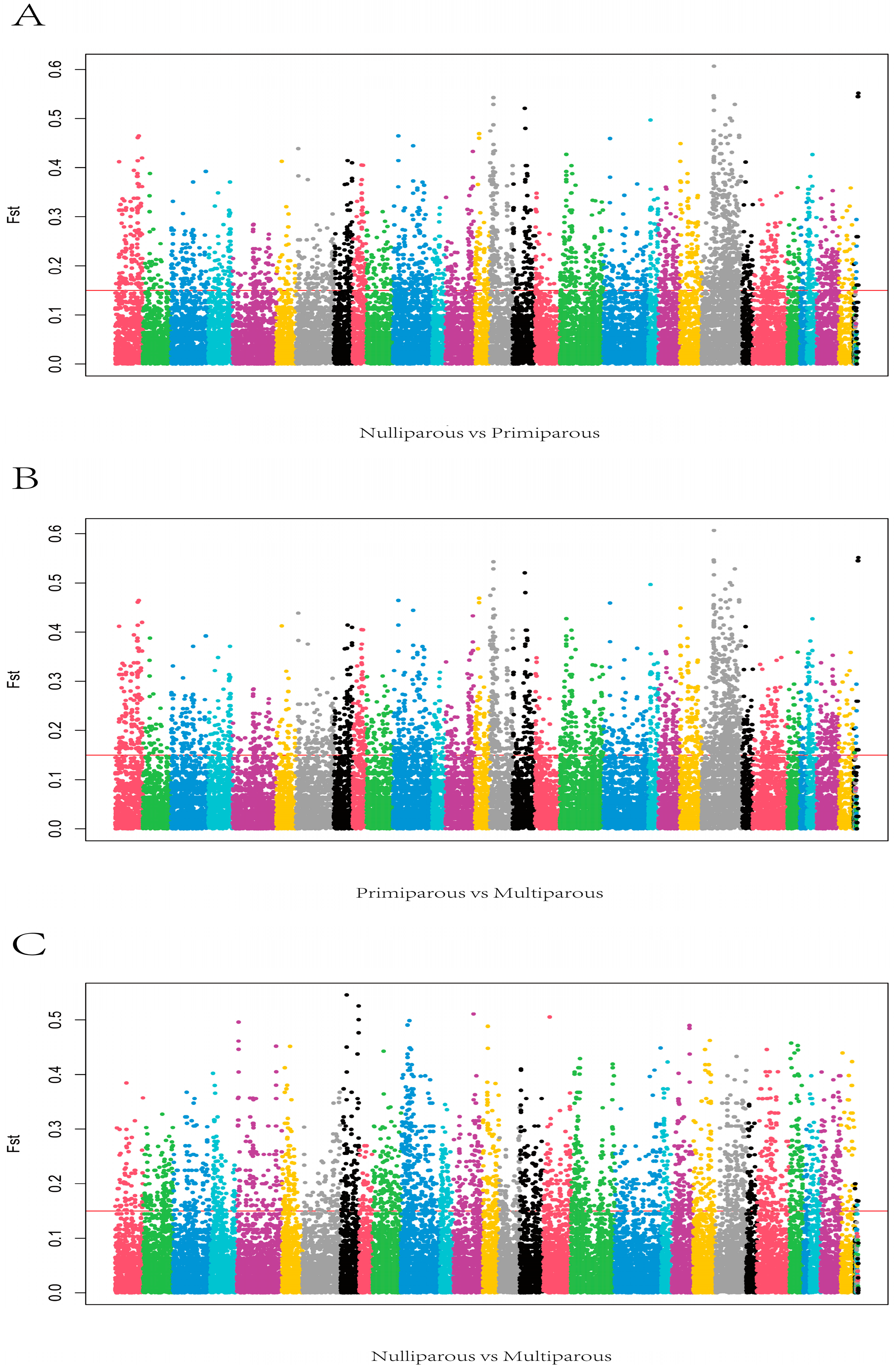
3.6. Exon Wide Selection Signature
3.7. Top Genes Expressed in Three Periods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barrier, A.C.; Haskell, M.J.; Birch, S.; Bagnall, A.; Bell, D.J.; Dickinson, J.; Macrae, A.I.; Dwyer, C.M. The Impact of Dystocia on Dairy Calf Health, Welfare, Performance and Survival. Vet. J. 2013, 195, 86–90. [Google Scholar] [CrossRef]
- Berry, D.; Conroy, S.; Pabiou, T.; Cromie, A. Animal breeding strategies can improve meat quality attributes within entire populations. Meat Sci. 2017, 132, 6–18. [Google Scholar] [CrossRef]
- Bengtsson, C.; Stålhammar, H.; Strandberg, E.; Eriksson, S.; Fikse, W. Association of genomically enhanced and parent average breeding values with cow performance in Nordic dairy cattle. J. Dairy Sci. 2020, 103, 6383–6391. [Google Scholar] [CrossRef]
- Berry, D.; Ring, S. Observed progeny performance validates the benefit of mating genetically elite beef sires to dairy females. J. Dairy Sci. 2020, 103, 2523–2533. [Google Scholar] [CrossRef]
- Braun, U.; Hilbe, M.; Peterhans, E.; Schweizer, M. Border disease in cattle. Vet. J. 2019, 246, 12–20. [Google Scholar] [CrossRef]
- Berry, D.P.; Evans, R.D. Genetics of reproductive performance in seasonal calving beef cows and its association with performance traits. J. Anim. Sci. 2014, 92, 1412–1422. [Google Scholar] [CrossRef]
- Inoue, K.; Inoue, Y.; Oe, T.; Nishimura, M. Genomic imprinting variances of beef carcass traits and physiochemical characteristics in Japanese Black cattle. Anim. Sci. J. 2021, 92, e13504. [Google Scholar] [CrossRef]
- Fodor, I.; Gábor, G.; Lang, Z.; Abonyi-Tóth, Z.; Ózsvári, L. Relationship between reproductive management practices and fertility in primiparous and multiparous dairy cows. Can. J. Vet. Res. Rev. Can. Rech. Vet. 2019, 83, 218–227. [Google Scholar]
- Reimus, K.; Orro, T.; Emanuelson, U.; Viltrop, A.; Mõtus, K. On-farm mortality and related risk factors in Estonian dairy cows. Prev. Vet. Med. 2018, 155, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Friggens, N.; Lucy, M.; Roche, J. Milk Production and Fertility in Cattle. Annu. Rev. Anim. Biosci. 2016, 4, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Bickett-Weddle, D.A.; Sanderson, M.W.; Parker, E.J. Foreign Animal Disease Outbreaks. Vet. Clin. N. Am. Food Anim. Pract. 2018, 34, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Ochsner, K.P.; MacNeil, M.D.; Lewis, R.M.; Spangler, M.L. Economic Selection Index Development for Beefmaster Cattle Ii: General-Purpose Breeding Objective. J. Anim. Sci. 2017, 95, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Twomey, A.J.; Cromie, A.R.; McHugh, N.; Berry, D.P. Validation of a beef cattle maternal breeding objective based on a cross-sectional analysis of a large national cattle database. J. Anim. Sci. 2020, 98, skaa322. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, P.T.; Houe, H. Cow mortality as an indicator of animal welfare in dairy herds. Res. Vet. Sci. 2018, 119, 239–243. [Google Scholar] [CrossRef]
- Hirooka, H. Marbled Japanese Black cattle. J. Anim. Breed. Genet. 2014, 131, 1–2. [Google Scholar] [CrossRef]
- Ueda, S.; Iwamoto, E.; Kato, Y.; Shinohara, M.; Shirai, Y.; Yamanoue, M. Comparative metabolomics of Japanese Black cattle beef and other meats using gas chromatography–mass spectrometry. Biosci. Biotechnol. Biochem. 2019, 83, 137–147. [Google Scholar] [CrossRef]
- Sasazaki, S. Development of DNA markers for improvement of meat quality in a Japanese Black cattle population in Hyogo Prefecture. Anim. Sci. J. 2021, 92, e13663. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, O.M.; Whitwham, A.; Keane, T.; McCarthy, A.S.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Zhao, S.; Ye, Z.; Stanton, R. Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols. RNA 2020, 26, 903–909. [Google Scholar] [CrossRef]
- Li, B.; Gong, Y.; Zhang, Y.; Kim, K.S.; Xiao, Y.; Zeng, Q.; Xu, K.; Duan, Y.; He, J.; Ma, H.; et al. Spatiotemporal Regulation of Circular RNA Expression During the Developmentof Skeletal Muscle, Subcutaneous Fat, and Liver in Ningxiang Pigs. Genes 2022, 13, 746. [Google Scholar] [CrossRef]
- Chen, C.; Rui, X.; Hao, C.; He, Y. Tbtools, a Toolkit for Biologists Integrating Various Hts-Data Handling Tools with a User-Friendly Interface. BioRxiv 2018, 289660. [Google Scholar]
- Zhang, T.; Zhang, X.; Han, K.; Zhang, G.; Wang, J.; Xie, K.; Xue, Q.; Fan, X. Analysis of long noncoding RNA and mRNA using RNA sequencing during the differentiation of intramuscular preadipocytes in chicken. PLoS ONE 2017, 12, e0172389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cai, W.; Li, Q.; Wang, Y.; Wang, Z.; Zhang, Q.; Xu, L.; Xu, L.; Hu, X.; Zhu, B.; et al. Transcriptome Analysis of Bovine Rumen Tissue in Three Developmental Stages. Front. Genet. 2022, 13, 821406. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, L.; Wang, J.; Zhang, M.; Song, Z.; Ni, B.; You, Y. Identification of Key Biomarkers and Immune Infiltration in Systemic Lupus Erythematosus by Integrated Bioinformatics Analysis. J. Transl. Med. 2021, 19, 35. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, P.; He, S.; Xing, S.; Cao, Z.; Cao, X.; Wang, X.; Li, Z.-H. Effects of short-term exposure to tralopyril on physiological indexes and endocrine function in turbot (Scophthalmus maximus). Aquat. Toxicol. 2022, 245. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Koh, J.-M.; Hwang, J.-Y.; Choi, K.-Y.; Lee, S.H.; Park, E.K.; Kim, T.-H.; Han, B.G.; Kim, G.S.; Kim, S.-Y.; et al. Association of a RUNX2 Promoter Polymorphism with Bone Mineral Density in Postmenopausal Korean Women. Calcif. Tissue Int. 2009, 84, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Guillocheau, G.M.; El Hou, A.; Meersseman, C.; Esquerré, D.; Rebours, E.; Letaief, R.; Simao, M.; Hypolite, N.; Bourneuf, E.; Bruneau, N.; et al. Survey of allele specific expression in bovine muscle. Sci. Rep. 2019, 9, 4297. [Google Scholar] [CrossRef]
- Lu, H.; Jiang, T.; Ren, K.; Li, Z.L.; Ren, J.; Wu, G.; Han, X. RUNX2 Plays An Oncogenic Role in Esophageal Carcinoma by Activating the PI3K/AKT and ERK Signaling Pathways. Cell. Physiol. Biochem. 2018, 49, 217–225. [Google Scholar] [CrossRef]
- Chamseddine, A.N.; Cabrero, M.; Wei, Y.; Ganan-Gomez, I.; Colla, S.; Takahashi, K.; Yang, H.; Bohannan, Z.S.; Garcia-Manero, G. PDE4 Differential Expression Is a Potential Prognostic Factor and Therapeutic Target in Patients with Myelodysplastic Syndrome and Chronic Myelomonocytic Leukemia. Clin. Lymphoma Myeloma Leuk. 2016, 16, S67–S73. [Google Scholar] [CrossRef] [PubMed]
- Schmetterer, K.G.; Goldhahn, K.; Ziegler, L.S.; Gerner, M.C.; Marculescu, R. Overexpression of Pde4a Acts as Checkpoint Inhibitor against Camp-Mediated Immunosuppression in Vitro. Front. Immunol. 2019, 10, 1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogan, S.; Houslay, M. Challenge of human Jurkat T-cells with the adenylate cyclase activator forskolin elicits major changes in cAMP phosphodiesterase (PDE) expression by up-regulating PDE3 and inducing PDE4D1 and PDE4D2 splice variants as well as down-regulating a novel PDE4A splice variant. Biochem. J. 1997, 321, 165–175. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Holl, K.; DeBehnke, S.; Yeo, C.T.; Hansen, P.; Gebre, A.K.; Leone-Kabler, S.; Ruas, M.; Parks, J.S.; Parrington, J. Genetics of Metabolic Syndrome: Tpcn2 Knockout Mice Have Improved Insulin Sensitivity and Are Protected against High-Fat Diet-Induced Weight Gain. Physiol. Genom. 2018, 50, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Schön, C.; Chen, C.-C.; Yang, Z.; Liegl, R.; Murenu, E.; Schworm, B.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; et al. TPC2 promotes choroidal angiogenesis and inflammation in a mouse model of neovascular age-related macular degeneration. Life Sci. Alliance 2021, 4, e202101047. [Google Scholar] [CrossRef]
- Urabe, F.; Kosaka, N.; Sawa, Y.; Yamamoto, Y.; Ito, K.; Yamamoto, T.; Kimura, T.; Egawa, S.; Ochiya, T. miR-26a regulates extracellular vesicle secretion from prostate cancer cells via targeting SHC4, PFDN4, and CHORDC1. Sci. Adv. 2020, 6, eaay3051. [Google Scholar] [CrossRef]
- Netcharoensirisuk, P.; Abrahamian, C.; Tang, R.; Chen, C.C.; Rosato, A.S.; Beyers, W.; Chao, Y.K.; Filippini, A.; Di Pietro, S.; Bartel, K.; et al. Flavonoids increase melanin production and reduce proliferation, migration and invasion of melanoma cells by blocking endolysosomal/melanosomal TPC2. Sci. Rep. 2021, 11, 8515. [Google Scholar] [CrossRef]
- Shihua, D.; Yanfeng, S. Microrna-22 May Promote Apoptosis and Inhibit the Proliferation of Hypertrophic Scar Fibroblasts by Regulating the Mitogen-Activated Protein Kinase Kinase/Extracellular Signal-Regulated Kinase/P21 Pathway. Experim. Therap. Med. 2017, 14, 3841–3845. [Google Scholar]
- Li, B.; Zhu, F.-C.; Yu, S.-X.; Liu, S.-J.; Li, B.-Y. Suppression of KIF22 Inhibits Cell Proliferation and Xenograft Tumor Growth in Colon Cancer. Cancer Biother. Radiopharm. 2020, 35, 50–57. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, F.; Yu, K.L.; Tas, R.; Grigoriev, I.; Remmelzwaal, S.; Serra-Marques, A.; Kapitein, L.C.; Heck, A.J.R.; Akhmanova, A. MICAL3 Flavoprotein Monooxygenase Forms a Complex with Centralspindlin and Regulates Cytokinesis. J. Biol. Chem. 2016, 291, 20617–20629. [Google Scholar] [CrossRef]
- Ehrlich, K.; Baribault, C.; Ehrlich, M. Epigenetics of Muscle- and Brain-Specific Expression of KLHL Family Genes. Int. J. Mol. Sci. 2020, 21, 8394. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, R.; Vathsala, V.; Sriram, T. Phenytoin Induced Down Regulation of Tesk2 Gene in Albino Rat Testis Gene Qrt PCR Analysis. J. Chem. Pharma. Sci. 2013, 6, 274–279. [Google Scholar]
- Watt, K.E.; Neben, C.L.; Hall, S.; Merrill, A.E.; Trainor, P.A. Tp53-Dependent and Independent Signaling Underlies the Pathogenesis and Possible Prevention of Acrofacial Dysostosis-Cincinnati Type. Human Mol. Genetics 2018, 27, 2628–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Chang, C.; Baiyin, B.; Liu, Z.; Guo, L.; Zhou, L.; Liu, B.; Shi, C.; Zhang, W. Blood Transcriptome Analysis of Beef Cow with Different Parity Revealed Candidate Genes and Gene Networks Regulating the Postpartum Diseases. Genes 2022, 13, 1671. https://doi.org/10.3390/genes13091671
Yang Y, Chang C, Baiyin B, Liu Z, Guo L, Zhou L, Liu B, Shi C, Zhang W. Blood Transcriptome Analysis of Beef Cow with Different Parity Revealed Candidate Genes and Gene Networks Regulating the Postpartum Diseases. Genes. 2022; 13(9):1671. https://doi.org/10.3390/genes13091671
Chicago/Turabian StyleYang, Yanda, Chencheng Chang, Batu Baiyin, Zaixia Liu, Lili Guo, Le Zhou, Bin Liu, Caixia Shi, and Wenguang Zhang. 2022. "Blood Transcriptome Analysis of Beef Cow with Different Parity Revealed Candidate Genes and Gene Networks Regulating the Postpartum Diseases" Genes 13, no. 9: 1671. https://doi.org/10.3390/genes13091671