
Assessment of Genetic Diversity, Runs of Homozygosity, and Signatures of Selection in Tropical Milking Criollo Cattle Using Pedigree and Genomic Data
 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Pedigree Analyses
2.2. Genomic Analyses
3. Results
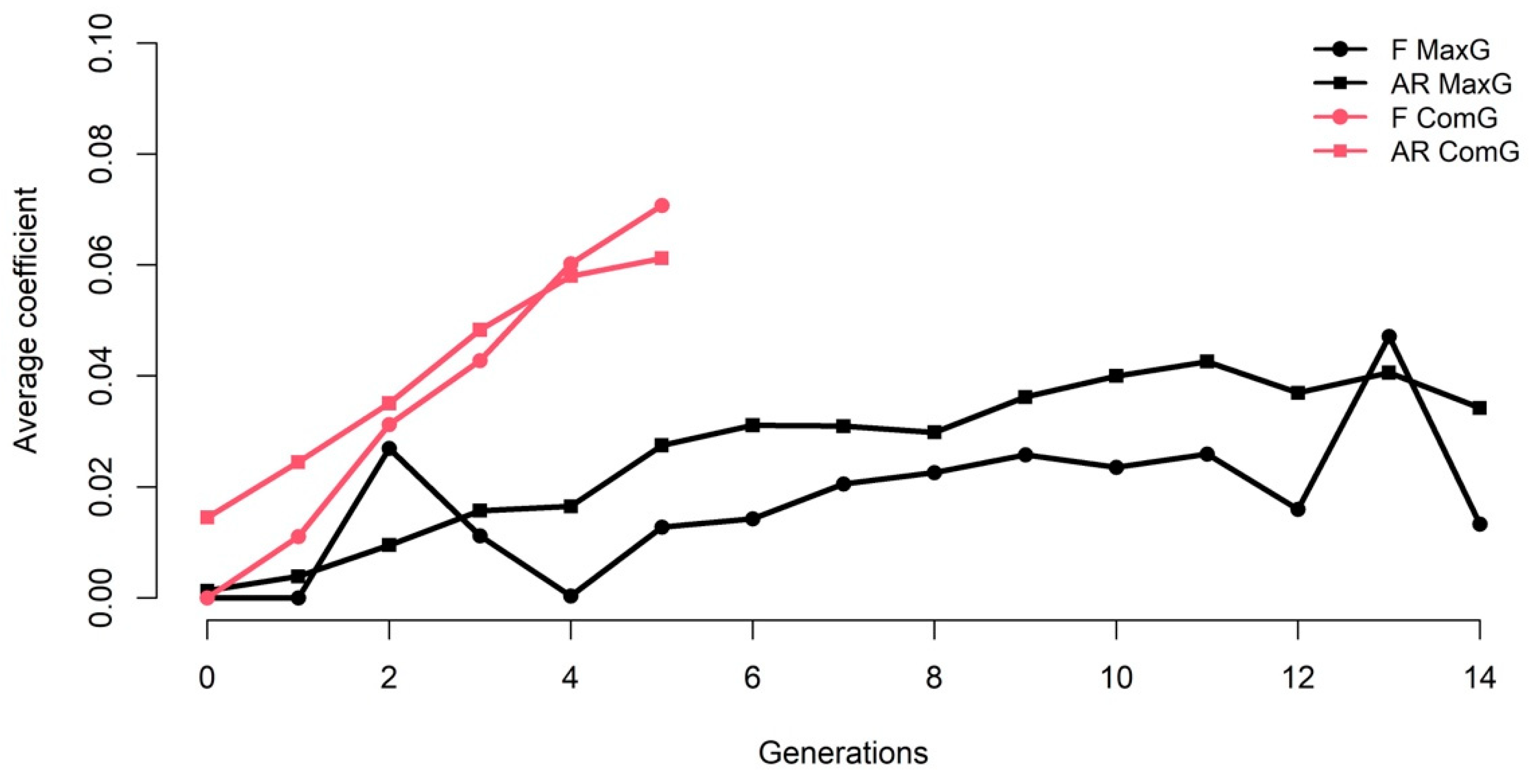
3.1. Pedigree Analyses
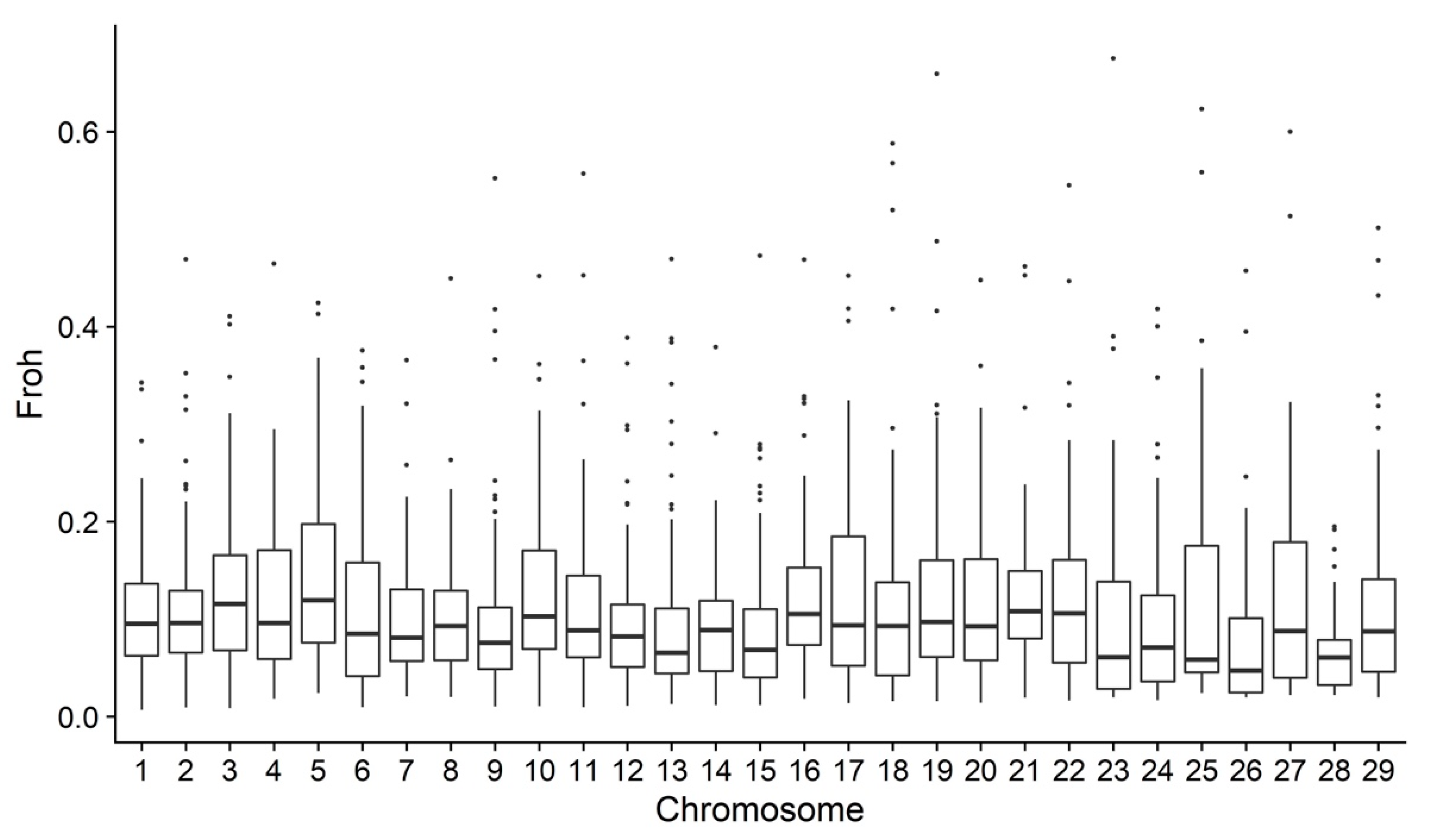
3.2. Genomic Analyses
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Groeneveld, L.F.; Lenstra, J.A.; Eding, H.; Toro, M.A.; Scherf, B.; Pilling, D.; Negrini, R.; Finlay, E.K.; Jianlin, H.; Groenevel, S.; et al. Genetic diversity in farm animals—A review. Amin. Genet. 2010, 41, 6–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, E.; Almeida, F.R.; McIntosh, M.M.; Poli, M.; Cibils, A.F.; Martínez-Quintana, J.A.; Félix-Portillo, M.; Estell, R.E. Genetic and productive background of Criollo cattle in Argentina, Mexico, Uruguay, and the United States. J. Arid Environ. 2022, 200, 104722. [Google Scholar] [CrossRef]
- De Alba, J.M. El Libro de los Bovinos Criollos en América; Mundi Prensa Mexico: Ciudad de México, México, 2011; p. 444. [Google Scholar]
- Rosendo-Ponce, A.; Becerril-Pérez, C.M. Avance en el conocimiento del bovino Criollo Lechero Tropical de México. Ecosistemas Y Recursos Agropecuarios 2015, 2, 233–243. [Google Scholar]
- Sheikhlou, M.; Abbasi, M.A. Genetic diversity of Iranian Lori-Bakhtiari sheep assessed by pedigree analysis. Small Rumin. Res. 2016, 141, 99–105. [Google Scholar] [CrossRef]
- Fabbri, M.C.; Dadousis, C.; Tiezzi, F.; Maltecca, C.; Lozada-Soto, E.; Biffani, S.; Bozzi, R. Genetic diversity and population history of eight Italian beef cattle breeds using measures of autozygosity. PLoS ONE 2021, 16, e0248087. [Google Scholar] [CrossRef]
- Hidalgo, J.; Cesarani, A.; Garcia, A.; Sumreddee, P.; Larios, N.; Mancin, E.; García, J.G.; Núñez, R.; Ramírez, R. Genetic Background and Inbreeding Depression in Romosinuano Cattle Breed in Mexico. Animals 2021, 11, 321. [Google Scholar] [CrossRef]
- Edea, Z.; Dessie, T.; Dadi, H.; Do, K.T.; Kim, K.S. Genetic diversity and population structure of Ethiopian sheep populations revealed by high-density SNP markers. Front. Genet. 2017, 8, 218. [Google Scholar] [CrossRef] [Green Version]
- Rosendo Ponce, A.; Jiménez, A.L.P.; Martínez, F.R.; Hernández, G.T.; Valverde, R.R.; Pérez, C.M.B. Genetic variability of Tropical Milking Criollo cattle of Mexico estimated from genealogy information. Rev. Col. Cien. Pec. 2018, 31, 196–203. [Google Scholar] [CrossRef]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species. BMC Genom. 2020, 21, 94. [Google Scholar] [CrossRef]
- Gutiérrez, J.P.; Goyache, F. A note on ENDOG: A computer program for analyzing pedigree information. J. Anim. Breed Genet. 2005, 122, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, T.H.E.; Luo, Z. Computing inbreeding coefficients in large populations. Genet. Sel. Evol. 1992, 24, 305–313. [Google Scholar] [CrossRef]
- Maignel, L.; Boichard, D.; Verrier, E. Genetic variability of French dairy breeds estimated from pedigree information. Interbull Bull. 1996, 14, 49. [Google Scholar]
- Lacy, R.C. Analysis of founder representation in pedigrees: Founder equivalents and founder genome equivalents. Zoo Biol. 1989, 8, 111–123. [Google Scholar] [CrossRef]
- Boichard, D.; Maignel, L.; Verrier, E. The value of using probabilities of gene origin to measure genetic variability in a population. Genet. Sel. Evol. 1997, 29, 1–19. [Google Scholar] [CrossRef]
- Gutiérrez, J.; Cervantes, I.; Goyache, F. Improving the estimation of realized effective population sizes in farm animals. J. Anim. Breed Genet. 2009, 126, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortellari, M.; Bionda, A.; Negro, A.; Frattini, S.; Mastrangelo, S.; Somenzi, E.; Lasagna, E.; Sarti, F.M.; Ciani, E.; Ciampolini, R.; et al. Runs of homozygosity in the Italian goat breeds: Impact of management practices in low-input systems. Genet. Sel. Evol. 2021, 53, 92. [Google Scholar] [CrossRef] [PubMed]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. DetectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes, 0.9.5. 2018. Available online: https://github.com/bioinformatics-ptp/detectRUNS/tree/master/detectRUNS (accessed on 25 July 2022).
- McQuillan, R.; Leutenegger, A.L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Qanbari, S.; Simianer, H. Mapping signatures of positive selection in the genome of livestock. Livest. Sci. 2014, 166, 133–143. [Google Scholar] [CrossRef]
- Zhang, Q.; Calus, M.P.; Bosse, M.; Sahana, G.; Lund, M.S.; Guldbrandtsen, B. Human-mediated introgression of haplotypes in a modern dairy cattle breed. Genetics 2018, 209, 1305–1317. [Google Scholar] [CrossRef] [Green Version]
- Cesarani, A.; Sorbolini, S.; Criscione, A.; Bordonaro, S.; Pulina, G.; Battacone, G.; Marletta, D.; Gaspa, G.; Macciotta, N.P.P. Genome-wide variability and selection signatures in Italian island cattle breeds. Amin. Genet. 2018, 49, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Franklin, I.R. Evolutionary change in small populations. In Conservation Biology: An Evolutionary-Ecological Perspective; Soulé, M.E., Wilcox, B.A., Eds.; Sinauer: Sunderland, MA, USA, 1980; pp. 135–149. [Google Scholar]
- FAO. Secondary Guidelines for Development of National Farm Animal Genetic Resources Management Plans: Management of Small Populations at Risk; FAO: Rome, Italy, 1998. [Google Scholar]
- Ramírez-Valverde, R.; Delgadillo-Zapata, A.R.; Domínguez-Viveros, J.; Hidalgo-Moreno, H.A.; Núñez-Domínguez, R.; Rodriguez-Almeida, F.A.; Reyes-Quiroz, C.; García-Muñiz, J.G. Pedigree analysis for determination of genetic diversity in Mexican beef cattle populations. Rev. Mex. Cienc. Pecu. 2018, 9, 614–635. [Google Scholar]
- Villanueva, B.; Fernández, A.; Saura, M.; Caballero, A.; Fernández, J.; Morales-Gonzáles, E.; Toro, M.A.; Pong-Wong, R. The value of genomic relationship matrices to estimate levels of inbreeding. Genet. Sel. Evol. 2021, 53, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Calus, M.P.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet. 2015, 16, 88. [Google Scholar] [CrossRef] [Green Version]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar] [CrossRef] [Green Version]
- Macciotta, N.P.P.; Colli, L.; Cesarani, A.; Ajmone-Marsan, P.; Low, W.Y.; Tearle, R.; Williams, J.L. The distribution of runs of homozygosity in the genome of river and swamp buffaloes reveals a history of adaptation, migration and crossbred events. Genet. Sel. Evol. 2021, 53, 20. [Google Scholar] [CrossRef]
- Nandolo, W.; Utsunomiya, Y.T.; Mészáros, G.; Wurzinger, M.; Khayadzadeh, N.; Torrecilha, R.B.P.; Mulindwa, H.A.; Gondwe, T.N.; Waldmann, P.; Ferenčaković, M.; et al. Misidentification of runs of homozygosity islands in cattle caused by interference with copy number variation or large intermarker distances. Genet. Sel. Evol. 2018, 50, 43. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, A.; Alijani, S.; Rafat, S.A.; Abdollahi-Arpanahi, R. Single-step genome-wide association study and candidate genes networks affecting reproductive traits in Iranian Holstein cattle. Livest. Sci. 2022, 262, 104971. [Google Scholar] [CrossRef]
- Wagner, P.; Yin, T.; Brügemann, K.; Engel, P.; Weimann, C.; Schlez, K.; König, S. Genome-wide associations for microscopic differential somatic cell count and specific mastitis pathogens in Holstein cows in compost-bedded pack and cubicle farming systems. Animals 2021, 11, 1839. [Google Scholar] [CrossRef]
- Ryu, J.; Lee, C. Genetic association of marbling score with intragenic nucleotide variants at selection signals of the bovine genome. Animal 2016, 10, 566–570. [Google Scholar] [CrossRef] [Green Version]
- Ramey, H.R.; Decker, J.E.; McKay, S.D.; Rolf, M.M.; Schnabel, R.D.; Taylor, J.F. Detection of selective sweeps in cattle using genome-wide SNP data. BMC Genom. 2013, 14, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flori, L.; Fritz, S.; Jaffrézic, F.; Boussaha, M.; Gut, I.; Heath, S.; Foulley, J.L.; Gautier, M. The genome response to artificial selection: A case study in dairy cattle. PLoS ONE 2009, 4, e6595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Kim, E.Y.; Seo, Y.M.; Yoon, T.K.; Lee, W.S.; Lee, K.A. Function of the pentose phosphate pathway and its key enzyme, transketolase, in the regulation of the meiotic cell cycle in oocytes. Clin. Exp. Reprod. Med. 2012, 39, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.J.; Burgoon, L.D.; Williams, K.J.; Forgacs, A.L.; Zacharewski, T.R. Comparative temporal and dosedependent morphological and transcriptional uterine effects elicited by tamoxifen and ethynylestradiol in immature, ovariectomized mice. BMC Genom. 2007, 8, 151. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Barhoumi, R.; Burghardt, R.; Liu, S.; Kim, K.; Safe, S. Molecular Mechanism of Inhibitory Aryl Hydrocarbon Receptor-Estrogen Receptor/Sp1 Cross Talk in Breast Cancer Cells. Mol. Endocrinol. 2006, 20, 2199–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spessotto, P.; Bulla, R.; Danussi, C.; Radillo, O.; Cervi, M.; Monami, G. EMILIN1 represents a major stromal element determining human trophoblast invasion of the uterine wall. J. Cell Sci. 2006, 119, 4574–4584. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Path of Selection | N | Average Generation Interval ± SE (y) |
|---|---|---|
| Sire–Sire | 187 | 7.45 ± 0.42 |
| Sire–Dam | 957 | 7.31 ± 0.16 |
| Dam–Sire | 183 | 6.94 ± 0.26 |
| Dam–Dam | 827 | 6.46 ± 0.12 |
| Total | 2154 | 6.97 ± 0.09 |
| FGRM | |||||||
|---|---|---|---|---|---|---|---|
| Mean ± SD (%) | −0.7 ± 3.8 | 10.9 ± 3.0 | 10.9 ± 3.0 | 6.0 ± 2.9 | 4.2 ± 2.6 | 3.1 ± 2.1 | 2.1 ± 1.1 |
| Min (%) | −14.9 | 5.3 | 5.7 | 0.8 | 0.16 | 0.3 | 0.6 |
| Max (%) | 25.9 | 18.0 | 18.7 | 13.1 | 11. | 8.4 | 5.2 |
| Mean Length ± SD 1 | |||
|---|---|---|---|
| ROH Length Class | Number of ROH | Mb | SNP |
| 1–2 Mb | 7236 | 1.35 ± 0.26 | 28 ± 17 |
| 2–4 Mb | 1444 | 2.66 ± 0.52 | 51 ± 28 |
| 4–8 Mb | 453 | 5.55 ± 1.16 | 123 ± 56 |
| 8–16 Mb | 268 | 11.28 ± 2.24 | 243 ± 70 |
| >16 Mb | 111 | 23.71 ± 7.21 | 465 ± 157 |
| Total | 9512 | 2.28 ± 3.06 | 48 ± 69 |
| 0.11 | 0.17 | 0.17 | 0.19 | 0.26 | 0.29 | 0.33 | ||
| ns | 0.10 | 0.10 | 0.15 | 0.23 | 0.24 | −0.02 | ||
| ns | ns | 1.00 | 0.96 | 0.91 | 0.85 | 0.62 | ||
| ns | ns | *** | 0.96 | 0.91 | 0.85 | 0.62 | ||
| ns | ns | *** | *** | 0.97 | 0.91 | 0.63 | ||
| * | ns | *** | *** | *** | 0.95 | 0.65 | ||
| ** | ns | *** | *** | *** | *** | 0.70 | ||
| ** | ns | *** | *** | *** | *** | *** |
| BTA | Start (bp) | End (bp) | n SNP | Genes |
|---|---|---|---|---|
| 11 | 71,623,956 | 72,732,727 | 17 | BABAM2, RBKS, MRPL33, SLC4A1AP, SUPT7L, GPN1, CCDC121, ZNF512, C11H2orf16, GCKR, FNDC4, IFT172, KRTCAP3, NRBP1, PPM1G, ZNF513, SNX17, EIF2B4, GTF3C2, MPV17, UCN, TRIM54, DNAJC5G, SLC30A3, CAD, ATRAID, SLC5A6, TCF23, PREB, ABHD1, CGREF1, KHK, EMILIN1, OST4, AGBL5, TRNAA-AGC, TRNAY-GUA, TMEM214, MAPRE3, DPYSL5 |
| 16 | 42,918,309 | 45,953,414 | 35 | PEX14, DFFA, CORT, CENPS, PGD, UBE4B, NMNAT1, CTNNBIP1, PIK3CD, TMEM201, SPSB1, MIR34A, CA6, ENO1, SLC45A1, LOC786597, ERRFI1, PARK7, TNFRSF9, VAMP3 |
| 21 | 31,679,297 | 46,217,299 | 41 | RCN2, PSTPIP1, TSPAN3, HMG20A, ODF3L1, CSPG4, SNX33, IMP3, SNUPN, PTPN9, MAN2C1, NEIL1, MIR631, COMMD4, PPCDC, SCAMP5, COX5A, FAM219B, MPI, SCAMP2, ULK3, CPLX3, CSK, EDC3, CLK3, ARID3B, UBL7, SEMA7A, CYP11A1, STRA6, ISLR, PML, STOML1, LOXL1, GZMB, STXBP6, MIR2888-1, NOVA1, G2E3, SCFD1, COCH, STRN3, AP4S1, NUBPL, AKAP6, MIR6522, EGLN3, SPTSSA, EAPP, SNX6, CFL2, BAZ1A, LOC613444, SRP54, FAM177A1, PPP2R3C, KIAA0391, PSMA6, NFKBIA, INSM2, BRMS1L |
| 22 | 49,273,889 | 49,841,675 | 12 | RBM15B, MANF, MAPKAPK3, CISH, HEMK1, C22H3orf18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Rocha, R.; Hidalgo, J.; Cesarani, A.; Ramírez-Valverde, R.; Núñez-Domínguez, R.; García-Muñiz, J.G.; Domínguez-Viveros, J. Assessment of Genetic Diversity, Runs of Homozygosity, and Signatures of Selection in Tropical Milking Criollo Cattle Using Pedigree and Genomic Data. Genes 2022, 13, 1896. https://doi.org/10.3390/genes13101896
Martínez-Rocha R, Hidalgo J, Cesarani A, Ramírez-Valverde R, Núñez-Domínguez R, García-Muñiz JG, Domínguez-Viveros J. Assessment of Genetic Diversity, Runs of Homozygosity, and Signatures of Selection in Tropical Milking Criollo Cattle Using Pedigree and Genomic Data. Genes. 2022; 13(10):1896. https://doi.org/10.3390/genes13101896
Chicago/Turabian StyleMartínez-Rocha, Ricardo, Jorge Hidalgo, Alberto Cesarani, Rodolfo Ramírez-Valverde, Rafael Núñez-Domínguez, José Guadalupe García-Muñiz, and Joel Domínguez-Viveros. 2022. "Assessment of Genetic Diversity, Runs of Homozygosity, and Signatures of Selection in Tropical Milking Criollo Cattle Using Pedigree and Genomic Data" Genes 13, no. 10: 1896. https://doi.org/10.3390/genes13101896