

SR Splicing Factors Promote Cancer via Multiple Regulatory Mechanisms
Abstract
:1. Introduction
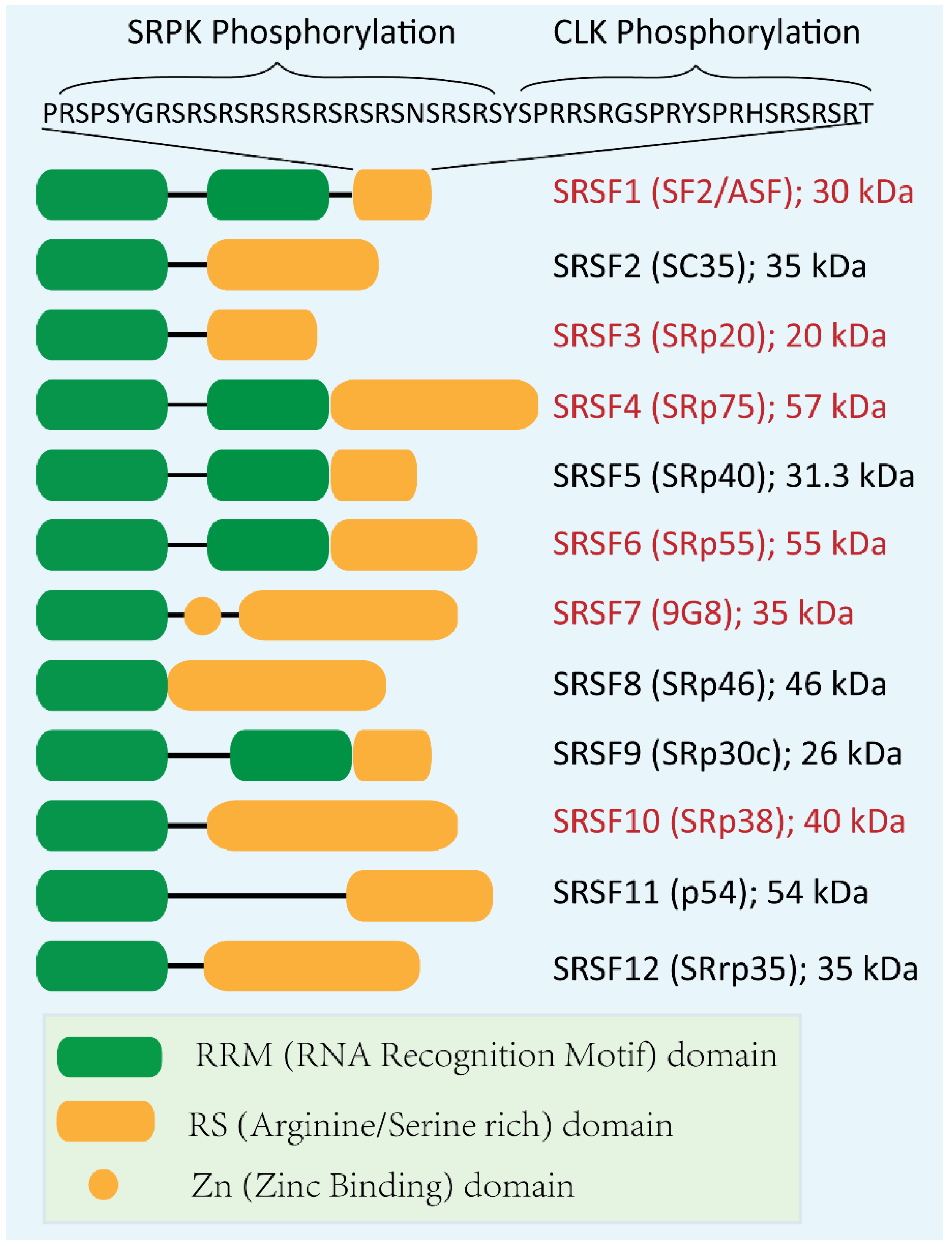
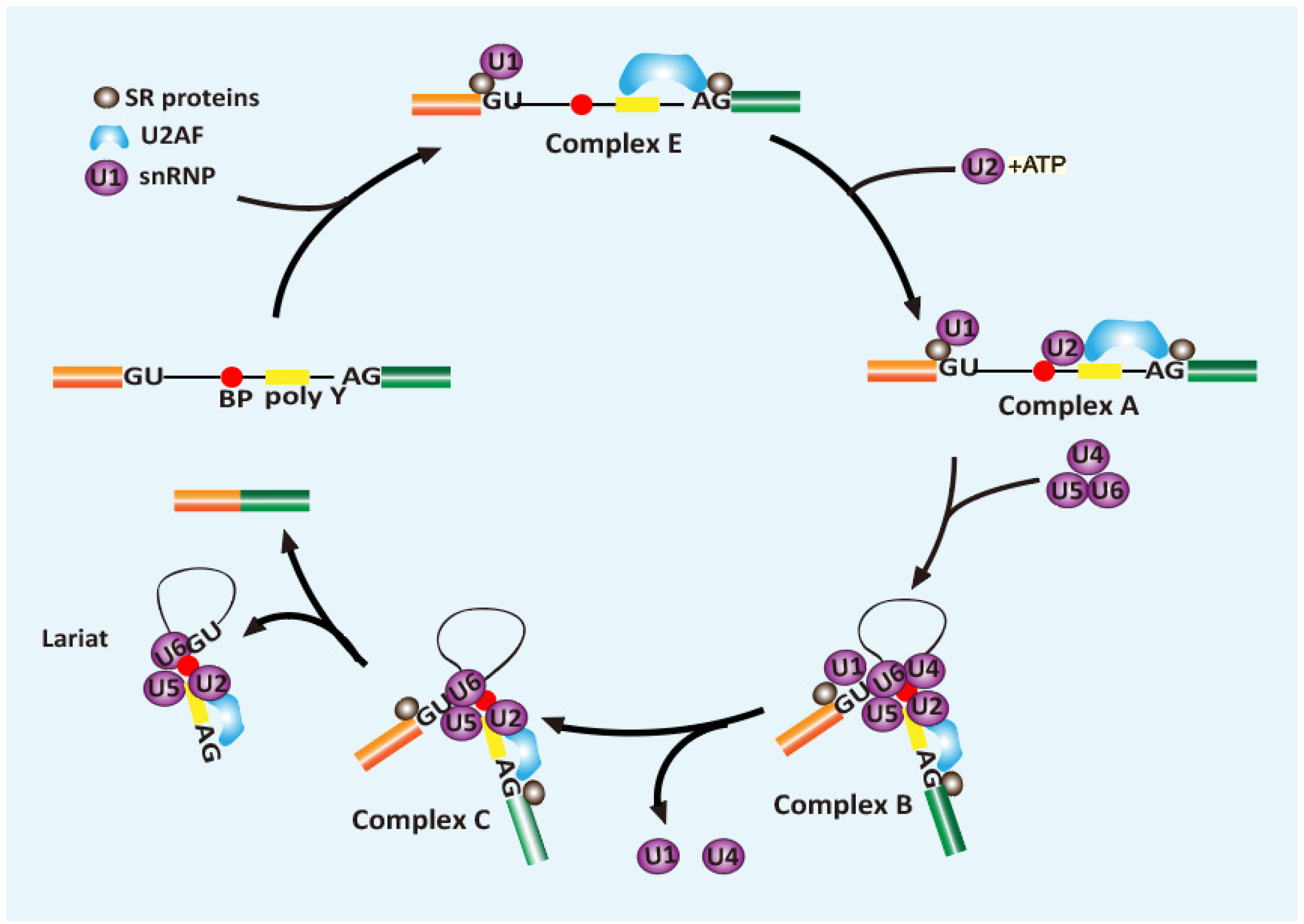
2. Role of SR Proteins in Splicing
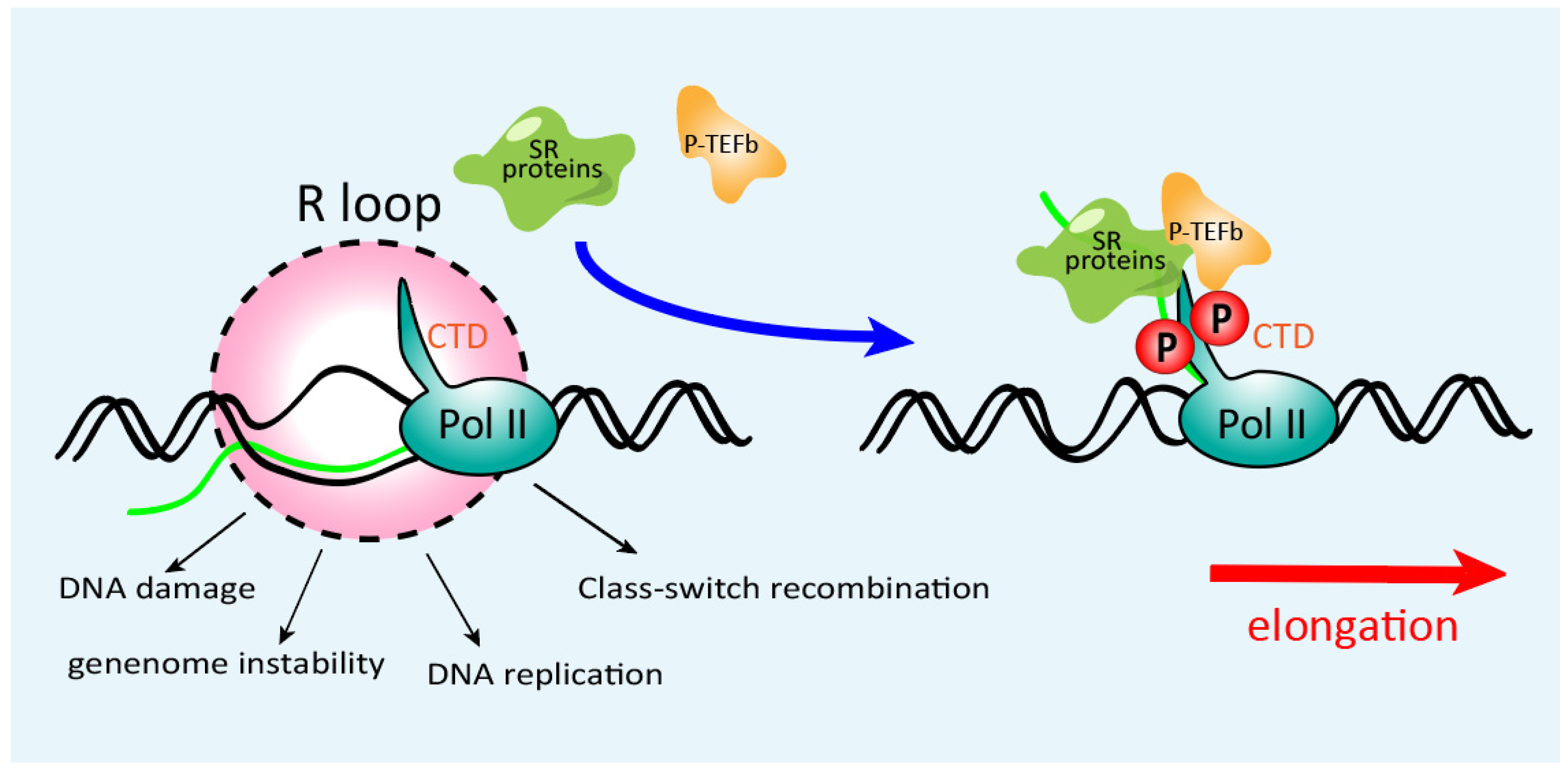
3. Role of SR Proteins in Transcriptional Elongation and Genomic Stability
4. Post-Splicing Activities of SR Proteins
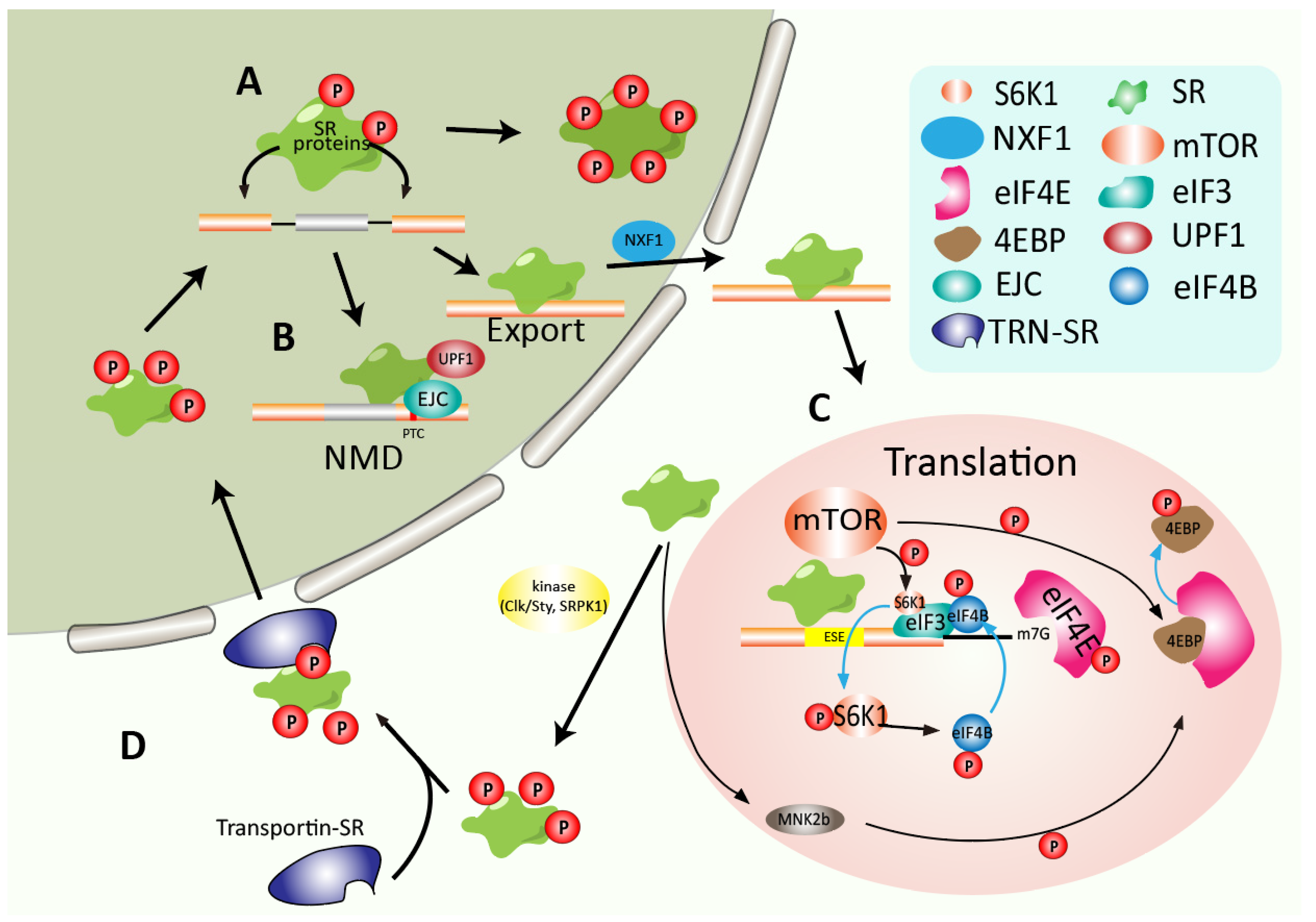
4.1. SR Proteins Promote Mature RNA Export from the Nucleus
4.2. Regulating mRNA Decay and Translation
5. Dysregulation of SR Proteins in Cancers
5.1. Aberrant SR Proteins Expression Is Associated with Cell Transformation and Tumorigenesis
5.2. Aberrant Phosphorylation of SR Protein in Cancer
5.3. SR proteins Link Alternative Splicing and Epigenetics Promoting Cancer Development
6. Competition or Coordination between Different SR Proteins
7. Targeting SR Proteins as Potential Cancer Therapeutics
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
- Gebauer, F.; Schwarzl, T.; Valcárcel, J.; Hentze, M.W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2021, 22, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Sanchez de Groot, N.; Armaos, A.; Graña-Montes, R.; Alriquet, M.; Calloni, G.; Vabulas, R.M.; Tartaglia, G.G. RNA structure drives interaction with proteins. Nat. Commun. 2019, 10, 3246. [Google Scholar] [CrossRef]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2008, 417, 15–27. [Google Scholar] [CrossRef]
- Rahman, M.A.; Krainer, A.R.; Abdel-Wahab, O. SnapShot: Splicing Alterations in Cancer. Cell 2020, 180, 208-208.e1. [Google Scholar] [CrossRef] [PubMed]
- Champlin, D.T.; Frasch, M.; Saumweber, H.; Lis, J.T. Characterization of a Drosophila protein associated with boundaries of transcriptionally active chromatin. Genes Dev. 1991, 5, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Manley, J.L. A protein factor, ASF, controls cell-specific alternative splicing of SV40 early pre-mRNA in vitro. Cell 1990, 62, 25–34. [Google Scholar] [CrossRef]
- Krainer, A.R.; Conway, G.C.; Kozak, D. The essential pre-mRNA splicing factor SF2 influences 5′ splice site selection by activating proximal sites. Cell 1990, 62, 35–42. [Google Scholar] [CrossRef]
- Leclair, N.K.; Brugiolo, M.; Urbanski, L.; Lawson, S.C.; Thakar, K.; Yurieva, M.; George, J.; Hinson, J.T.; Cheng, A.; Graveley, B.R.; et al. Poison Exon Splicing Regulates a Coordinated Network of SR Protein Expression during Differentiation and Tumorigenesis. Mol. Cell 2020, 80, 648–665.e649. [Google Scholar] [CrossRef]
- Kraus, M.E.; Lis, J.T. The concentration of B52, an essential splicing factor and regulator of splice site choice in vitro, is critical for Drosophila development. Mol. Cell. Biol. 1994, 14, 5360–5370. [Google Scholar]
- Jeong, S. SR Proteins: Binders, Regulators, and Connectors of RNA. Mol. Cells 2017, 40, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Manley, J.L. Inactivation of the SR Protein Splicing Factor ASF/SF2 Results in Genomic Instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Gattoni, R.; Stévenin, J.; Steitz, J.A. SR Splicing Factors Serve as Adapter Proteins for TAP-Dependent mRNA Export. Mol. Cell 2003, 11, 837–843. [Google Scholar] [CrossRef]
- Wagner, R.E.; Frye, M. Noncanonical functions of the serine-arginine-rich splicing factor (SR) family of proteins in development and disease. Bioessays 2021, 43, 2000242. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Angarola, B.L.; Anczuków, O. Splicing alterations in healthy aging and disease. Wiley Interdiscip. Rev. RNA 2021, 12, e1643. [Google Scholar] [CrossRef]
- Hegele, A.; Kamburov, A.; Grossmann, A.; Sourlis, C.; Wowro, S.; Weimann, M.; Will, C.L.; Pena, V.; Lührmann, R.; Stelzl, U. Dynamic Protein-Protein Interaction Wiring of the Human Spliceosome. Mol. Cell 2012, 45, 567–580. [Google Scholar] [CrossRef]
- Sztacho, M.; Šalovská, B.; Červenka, J.; Balaban, C.; Hoboth, P.; Hozák, P. Limited Proteolysis-Coupled Mass Spectrometry Identifies Phosphatidylinositol 4,5-Bisphosphate Effectors in Human Nuclear Proteome. Cells 2021, 10, 68. [Google Scholar] [CrossRef]
- Wilkinson, M.E.; Charenton, C.; Nagai, K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. [Google Scholar] [CrossRef]
- Ma, X.; He, F. Advances in the Study of SR Protein Family. Genom. Proteom. Bioinform. 2003, 1, 2–8. [Google Scholar] [CrossRef]
- Petermann, E.; Lan, L.; Zou, L. Sources, resolution and physiological relevance of R-loops and RNA–DNA hybrids. Nat. Rev. Mol. Cell Biol. 2022, 23, 521–540. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, J.; Leela, J.K.; Anupama, K. R-loops in bacterial transcription. Transcription 2013, 4, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Zatreanu, D.; Han, Z.; Mitter, R.; Tumini, E.; Williams, H.; Gregersen, L.; Dirac-Svejstrup, A.B.; Roma, S.; Stewart, A.; Aguilera, A.; et al. Elongation Factor TFIIS Prevents Transcription Stress and R-Loop Accumulation to Maintain Genome Stability. Mol. Cell 2019, 76, 57–69.e9. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 2014, 15, 163–175. [Google Scholar] [CrossRef]
- Saldi, T.; Cortazar, M.A.; Sheridan, R.M.; Bentley, D.L. Coupling of RNA Polymerase II Transcription Elongation with Pre-mRNA Splicing. J. Mol. Biol. 2016, 428, 2623–2635. [Google Scholar] [CrossRef]
- Drexler, H.L.; Choquet, K.; Churchman, L.S. Splicing Kinetics and Coordination Revealed by Direct Nascent RNA Sequencing through Nanopores. Mol. Cell 2020, 77, 985–998.e8. [Google Scholar] [CrossRef]
- Das, R.; Yu, J.; Zhang, Z.; Gygi, M.P.; Krainer, A.R.; Gygi, S.P.; Reed, R. SR Proteins Function in Coupling RNAP II Transcription to Pre-mRNA Splicing. Mol. Cell 2007, 26, 867–881. [Google Scholar] [CrossRef]
- Muniz, L.; Nicolas, E.; Trouche, D. RNA polymerase II speed: A key player in controlling and adapting transcriptome composition. EMBO J. 2021, 40, e105740. [Google Scholar] [CrossRef]
- Lin, S.; Coutinho-Mansfield, G.; Wang, D.; Pandit, S.; Fu, X.-D. The splicing factor SC35 has an active role in transcriptional elongation. Nat. Struct. Mol. Biol. 2008, 15, 819–826. [Google Scholar] [CrossRef]
- Xiao, R.; Sun, Y.; Ding, J.-H.; Lin, S.; Rose, D.W.; Rosenfeld, M.G.; Fu, X.-D.; Li, X. Splicing Regulator SC35 Is Essential for Genomic Stability and Cell Proliferation during Mammalian Organogenesis. Mol. Cell. Biol. 2007, 27, 5393–5402. [Google Scholar] [CrossRef]
- Müller-McNicoll, M.; Botti, V.; de Jesus Domingues, A.M.; Brandl, H.; Schwich, O.D.; Steiner, M.C.; Curk, T.; Poser, I.; Zarnack, K.; Neugebauer, K.M. SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev. 2016, 30, 553–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tretyakova, I.; Zolotukhin, A.S.; Tan, W.; Bear, J.; Propst, F.; Ruthel, G.; Felber, B.K. Nuclear Export Factor Family Protein Participates in Cytoplasmic mRNA Trafficking. J. Biol. Chem. 2005, 280, 31981–31990. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Lee, E.S. Sequence determinants for nuclear retention and cytoplasmic export of mRNAs and lncRNAs. Front. Genet. 2018, 9, 440. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Connections between the processing and nuclear export of mRNA: Evidence for an export license? Proc. Natl. Acad. Sci. USA 2000, 97, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Cáceres, J.F.; Screaton, G.R.; Krainer, A.R. A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev. 1998, 12, 55–66. [Google Scholar] [CrossRef]
- Sato, H.; Singer, R.H. Cellular variability of nonsense-mediated mRNA decay. Nat. Commun. 2021, 12, 7203. [Google Scholar] [CrossRef]
- Singh, G.; Kucukural, A.; Cenik, C.; Leszyk, J.D.; Shaffer, S.A.; Weng, Z.; Moore, M.J. The Cellular EJC Interactome Reveals Higher Order mRNP Structure and an EJC-SR Protein Nexus. Cell 2012, 151, 750–764. [Google Scholar] [CrossRef]
- Zhang, Z.; Krainer, A.R. Involvement of SR Proteins in mRNA Surveillance. Mol. Cell 2004, 16, 597–607. [Google Scholar] [CrossRef]
- Aznarez, I.; Nomakuchi, T.T.; Tetenbaum-Novatt, J.; Rahman, M.A.; Fregoso, O.; Rees, H.; Krainer, A.R. Mechanism of Nonsense-Mediated mRNA Decay Stimulation by Splicing Factor SRSF1. Cell Rep. 2018, 23, 2186–2198. [Google Scholar] [CrossRef]
- Sato, H.; Hosoda, N.; Maquat, L.E. Efficiency of the Pioneer Round of Translation Affects the Cellular Site of Nonsense-Mediated mRNA Decay. Mol. Cell 2008, 29, 255–262. [Google Scholar] [CrossRef]
- Sanford, J.R.; Gray, N.K.; Beckmann, K.; Cáceres, J.F. A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 2004, 18, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslon, M.M.; Heras, S.R.; Bellora, N.; Eyras, E.; Cáceres, J.F. The translational landscape of the splicing factor SRSF1 and its role in mitosis. Elife 2014, 3, e02028. [Google Scholar] [CrossRef]
- Swartz, J.E.; Bor, Y.-C.; Misawa, Y.; Rekosh, D.; Hammarskjold, M.-L. The Shuttling SR Protein 9G8 Plays a Role in Translation of Unspliced mRNA Containing a Constitutive Transport Element. J. Biol. Chem. 2007, 282, 19844–19853. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.M.; Daijogo, S.; Semler, B.L. A nucleo-cytoplasmic SR protein functions in viral IRES-mediated translation initiation. EMBO J. 2007, 26, 459–467. [Google Scholar] [CrossRef]
- Jiang, L.; Huang, J.; Higgs, B.W.; Hu, Z.; Xiao, Z.; Yao, X.; Conley, S.; Zhong, H.; Liu, Z.; Brohawn, P.; et al. Genomic Landscape Survey Identifies SRSF1 as a Key Oncodriver in Small Cell Lung Cancer. PLoS Genet. 2016, 12, e1005895. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Akerman, M.; Cléry, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-regulated alternative splicing in breast cancer. Mol. Cell 2015, 60, 105–117. [Google Scholar] [CrossRef]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef]
- Wan, L.; Yu, W.; Shen, E.; Sun, W.; Liu, Y.; Kong, J.; Wu, Y.; Han, F.; Zhang, L.; Yu, T.; et al. SRSF6-regulated alternative splicing that promotes tumour progression offers a therapy target for colorectal cancer. Gut 2019, 68, 118–129. [Google Scholar] [CrossRef]
- Du, J.-X.; Luo, Y.-H.; Zhang, S.-J.; Wang, B.; Chen, C.; Zhu, G.-Q.; Zhu, P.; Cai, C.-Z.; Wan, J.-L.; Cai, J.-L.; et al. Splicing factor SRSF1 promotes breast cancer progression via oncogenic splice switching of PTPMT1. J. Exp. Clin. Cancer Res. 2021, 40, 171. [Google Scholar] [CrossRef]
- Liu, J.; Huang, B.; Xiao, Y.; Xiong, H.M.; Li, J.; Feng, D.Q.; Chen, X.M.; Zhang, H.B.; Wang, X.Z. Aberrant Expression of Splicing Factors in Newly Diagnosed Acute Myeloid Leukemia. Oncol. Res. Treat. 2012, 35, 335–340. [Google Scholar] [CrossRef]
- Sinnakannu, J.R.; Lee, K.L.; Cheng, S.; Li, J.; Yu, M.; Tan, S.P.; Ong, C.C.H.; Li, H.; Than, H.; Anczuków-Camarda, O.; et al. SRSF1 mediates cytokine-induced impaired imatinib sensitivity in chronic myeloid leukemia. Leukemia 2020, 34, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Zhang, H.; Du, C.; Liu, X.; Zhu, S.; Zhang, W.; Li, Z.; Gao, C.; Zhao, X.; Mei, M.; et al. Correlation of SRSF1 and PRMT1 expression with clinical status of pediatric acute lymphoblastic leukemia. J. Hematol. Oncol. 2012, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Park, W.C.; Kim, H.-R.; Kang, D.B.; Ryu, J.-S.; Choi, K.-H.; Lee, G.-O.; Yun, K.J.; Kim, K.Y.; Park, R.; Yoon, K.-H.; et al. Comparative expression patterns and diagnostic efficacies of SR splicing factors and HNRNPA1 in gastric and colorectal cancer. BMC Cancer 2016, 16, 358. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Zhao, Q.; Zhao, J.; Zhang, W.; Sun, Y.; Qin, P.; Lv, Y.; Bai, L.; Yang, Q.; Chen, L.; et al. SRSF1 modulates PTPMT1 alternative splicing to regulate lung cancer cell radioresistance. EBioMedicine 2018, 38, 113–126. [Google Scholar] [CrossRef]
- Lv, Y.; Zhang, W.; Zhao, J.; Sun, B.; Qi, Y.; Ji, H.; Chen, C.; Zhang, J.; Sheng, J.; Wang, T.; et al. SRSF1 inhibits autophagy through regulating Bcl-x splicing and interacting with PIK3C3 in lung cancer. Signal Transduct. Target. Ther. 2021, 6, 108. [Google Scholar] [CrossRef]
- Shultz, J.C.; Goehe, R.W.; Wijesinghe, D.S.; Murudkar, C.; Hawkins, A.J.; Shay, J.W.; Minna, J.D.; Chalfant, C.E. Alternative Splicing of Caspase 9 Is Modulated by the Phosphoinositide 3-Kinase/Akt Pathway via Phosphorylation of SRp30a. Cancer Res. 2010, 70, 9185–9196. [Google Scholar] [CrossRef]
- Chen, L.; Luo, C.; Shen, L.; Liu, Y.; Wang, Q.; Zhang, C.; Guo, R.; Zhang, Y.; Xie, Z.; Wei, N.; et al. SRSF1 Prevents DNA Damage and Promotes Tumorigenesis through Regulation of DBF4B Pre-mRNA Splicing. Cell Rep. 2017, 21, 3406–3413. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, R.; Li, X.; Yu, L.; Hua, D.; Sun, C.; Shi, C.; Luo, W.; Rao, C.; Jiang, Z.; et al. Splicing factor SRSF1 promotes gliomagenesis via oncogenic splice-switching of MYO1B. J. Clin. Investig. 2019, 129, 676–693. [Google Scholar] [CrossRef]
- Sokół, E.; Kędzierska, H.; Czubaty, A.; Rybicka, B.; Rodzik, K.; Tański, Z.; Bogusławska, J.; Piekiełko-Witkowska, A. microRNA-mediated regulation of splicing factors SRSF1, SRSF2 and hnRNP A1 in context of their alternatively spliced 3′UTRs. Exp. Cell Res. 2018, 363, 208–217. [Google Scholar] [CrossRef]
- Abou Faycal, C.; Gazzeri, S.; Eymin, B. A VEGF-A/SOX2/SRSF2 network controls VEGFR1 pre-mRNA alternative splicing in lung carcinoma cells. Sci. Rep. 2019, 9, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, C.; Cheng, Y.; Liu, Y.; Chen, L.; Liu, L.; Wei, N.; Xie, Z.; Wu, W.; Feng, Y. SRSF2 Regulates Alternative Splicing to Drive Hepatocellular Carcinoma Development. Cancer Res. 2017, 77, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Arslan, A.D.; Pool, M.D.; Ho, T.T.; Darcy, K.M.; Coon, J.S.; Beck, W.T. Knockdown of splicing factor SRp20 causes apoptosis in ovarian cancer cells and its expression is associated with malignancy of epithelial ovarian cancer. Oncogene 2011, 30, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, M.; Meng, F.; Zhang, Y.; Wang, M.; Guo, X.; Yang, J.; Zhang, H.; Zhang, H.; Sun, J.; et al. SRSF3 Promotes Angiogenesis in Colorectal Cancer by Splicing SRF. Front. Oncol. 2022, 12, 270. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Guo, J.; Jia, R. Oncogene SRSF3 suppresses autophagy via inhibiting BECN1 expression. Biochem. Biophys. Res. Commun. 2019, 509, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wan, X.; Huang, T.; Zeng, C.; Sastry, N.; Wu, B.; James, C.D.; Horbinski, C.; Nakano, I.; Zhang, W.; et al. SRSF3-Regulated RNA Alternative Splicing Promotes Glioblastoma Tumorigenicity by Affecting Multiple Cellular Processes. Cancer Res. 2019, 79, 5288–5301. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, C.; Wang, Z.-W.; Hu, J.-F.; Pan, J.-J.; Liao, C.-Y.; Zhang, J.-Q.; Chen, J.-Z.; Huang, Y.; Huang, L.; et al. CLK1/SRSF5 pathway induces aberrant exon skipping of METTL14 and Cyclin L2 and promotes growth and metastasis of pancreatic cancer. J. Hematol. Oncol. 2021, 14, 60. [Google Scholar] [CrossRef]
- Gautrey, H.L.; Tyson-Capper, A.J. Regulation of Mcl-1 by SRSF1 and SRSF5 in Cancer Cells. PLoS ONE 2012, 7, e51497. [Google Scholar] [CrossRef]
- Cohen-Eliav, M.; Golan-Gerstl, R.; Siegfried, Z.; Andersen, C.L.; Thorsen, K.; Ørntoft, T.F.; Mu, D.; Karni, R. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J. Pathol. 2013, 229, 630–639. [Google Scholar] [CrossRef]
- Choi, N.; Jang, H.N.; Oh, J.; Ha, J.; Park, H.; Zheng, X.; Lee, S.; Shen, H. SRSF6 Regulates the Alternative Splicing of the Apoptotic Fas Gene by Targeting a Novel RNA Sequence. Cancers 2022, 14, 1990. [Google Scholar] [CrossRef]
- Fu, Y.; Wang, Y. SRSF7 knockdown promotes apoptosis of colon and lung cancer cells. Oncol. Lett. 2018, 15, 5545–5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saijo, S.; Kuwano, Y.; Masuda, K.; Nishikawa, T.; Rokutan, K.; Nishida, K. Serine/arginine-rich splicing factor 7 regulates p21-dependent growth arrest in colon cancer cells. J. Med. Investig. 2016, 63, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, X.; Wang, P.; Chen, Q.; Xiong, L.; Tang, M.; Hong, C.; Lin, X.; Shi, K.; Liang, L.; et al. SRSF9 promotes colorectal cancer progression via stabilizing DSN1 mRNA in an m6A-related manner. J. Transl. Med. 2022, 20, 198. [Google Scholar] [CrossRef]
- Wang, R.; Su, Q.; Yin, H.; Wu, D.; Lv, C.; Yan, Z. Inhibition of SRSF9 enhances the sensitivity of colorectal cancer to erastin-induced ferroptosis by reducing glutathione peroxidase 4 expression. Int. J. Biochem. Cell Biol. 2021, 134, 105948. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Dai, M.; Xu, Q.; Zhu, X.; Zhou, Y.; Jiang, S.; Wang, Y.; Ai, Z.; Ma, L.; Zhang, Y.; et al. SRSF10-mediated IL1RAP alternative splicing regulates cervical cancer oncogenesis via mIL1RAP-NF-κB-CD47 axis. Oncogene 2018, 37, 2394–2409. [Google Scholar] [CrossRef]
- Chang, C.; Rajasekaran, M.; Qiao, Y.; Dong, H.; Wang, Y.; Xia, H.; Deivasigamani, A.; Wu, M.; Sekar, K.; Gao, H.; et al. The aberrant upregulation of exon 10-inclusive SREK1 through SRSF10 acts as an oncogenic driver in human hepatocellular carcinoma. Nat. Commun. 2022, 13, 1363. [Google Scholar] [CrossRef]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Das, S.; Anczuków, O.; Akerman, M.; Krainer, A.R. Oncogenic Splicing Factor SRSF1 Is a Critical Transcriptional Target of MYC. Cell Rep. 2012, 1, 110–117. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, Z.; Sinha, R.; Karni, R.; Krainer, A.R. SF2/ASF autoregulation involves multiple layers of post-transcriptional and translational control. Nat. Struct. Mol. Biol. 2010, 17, 306–312. [Google Scholar] [CrossRef]
- Fischer, D.-C.; Noack, K.; Runnebaum, I.B.; Watermann, D.O.; Kieback, D.G.; Stamm, S.; Stickeler, E. Expression of splicing factors in human ovarian cancer. Oncol. Rep. 2004, 11, 1085–1090. [Google Scholar] [CrossRef]
- Fu, Y.; Huang, B.; Shi, Z.; Han, J.; Wang, Y.; Huangfu, J.; Wu, W. SRSF1 and SRSF9 RNA binding proteins promote Wnt signalling-mediated tumorigenesis by enhancing β-catenin biosynthesis. EMBO Mol. Med. 2013, 5, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The regulation of protein function by multisite phosphorylation—A 25 year update. Trends Biochem. Sci. 2000, 25, 596–601. [Google Scholar] [CrossRef]
- Hamelberg, D.; Shen, T.; McCammon, J.A. A proposed signaling motif for nuclear import in mRNA processing via the formation of arginine claw. Proc. Natl. Acad. Sci. USA 2007, 104, 14947–14951. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.-C.; Lin, R.-I.; Tarn, W.-Y. Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 10154–10159. [Google Scholar] [CrossRef]
- Xiao, S.H.; Manley, J.L. Phosphorylation of the ASF/SF2 RS domain affects both protein-protein and protein-RNA interactions and is necessary for splicing. Genes Dev. 1997, 11, 334–344. [Google Scholar] [CrossRef]
- Huang, Y.; Yario, T.A.; Steitz, J.A. A molecular link between SR protein dephosphorylation and mRNA export. Proc. Natl. Acad. Sci. USA 2004, 101, 9666–9670. [Google Scholar] [CrossRef]
- Velazquez-Dones, A.; Hagopian, J.C.; Ma, C.-T.; Zhong, X.-Y.; Zhou, H.; Ghosh, G.; Fu, X.-D.; Adams, J.A. Mass Spectrometric and Kinetic Analysis of ASF/SF2 Phosphorylation by SRPK1 and Clk/Sty. J. Biol. Chem. 2005, 280, 41761–41768. [Google Scholar] [CrossRef]
- Serrano, P.; Aubol, B.E.; Keshwani, M.M.; Forli, S.; Ma, C.-T.; Dutta, S.K.; Geralt, M.; Wüthrich, K.; Adams, J.A. Directional Phosphorylation and Nuclear Transport of the Splicing Factor SRSF1 Is Regulated by an RNA Recognition Motif. J. Mol. Biol. 2016, 428, 2430–2445. [Google Scholar] [CrossRef]
- Wu, Q.; Chang, Y.; Zhang, L.; Zhang, Y.; Tian, T.; Feng, G.; Zhou, S.; Zheng, Q.; Han, F.; Huang, F. SRPK1 Dissimilarly Impacts on the Growth, Metastasis, Chemosensitivity and Angiogenesis of Glioma in Normoxic and Hypoxic Conditions. J. Cancer 2013, 4, 727–735. [Google Scholar] [CrossRef]
- van Roosmalen, W.; Le Dévédec, S.E.; Golani, O.; Smid, M.; Pulyakhina, I.; Timmermans, A.M.; Look, M.P.; Zi, D.; Pont, C.; de Graauw, M.; et al. Tumor cell migration screen identifies SRPK1 as breast cancer metastasis determinant. J. Clin. Investig. 2015, 125, 1648–1664. [Google Scholar] [CrossRef]
- Mavrou, A.; Brakspear, K.; Hamdollah-Zadeh, M.; Damodaran, G.; Babaei-Jadidi, R.; Oxley, J.; Gillatt, D.A.; Ladomery, M.R.; Harper, S.J.; Bates, D.O.; et al. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 2015, 34, 4311–4319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksaas, A.K.; Eikvar, S.; Akusjärvi, G.; Skålhegg, B.S.; Kvissel, A.K. Protein Kinase A–Dependent Phosphorylation of Serine 119 in the Proto-Oncogenic Serine/Arginine-Rich Splicing Factor 1 Modulates Its Activity as a Splicing Enhancer Protein. Genes Cancer 2011, 2, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Naviglio, S.; Caraglia, M.; Abbruzzese, A.; Chiosi, E.; Di Gesto, D.; Marra, M.; Romano, M.; Sorrentino, A.; Sorvillo, L.; Spina, A.; et al. Protein kinase A as a biological target in cancer therapy. Expert Opin. Ther. Targets 2009, 13, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Gardner, L.; Corn, P.G. Hypoxic regulation of mRNA expression. Cell Cycle 2008, 7, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Yan, Q. The roles of epigenetics in cancer progression and metastasis. Biochem. J. 2021, 478, 3373–3393. [Google Scholar] [CrossRef]
- Yearim, A.; Gelfman, S.; Shayevitch, R.; Melcer, S.; Glaich, O.; Mallm, J.-P.; Nissim-Rafinia, M.; Cohen, A.-H.S.; Rippe, K.; Meshorer, E.; et al. HP1 Is Involved in Regulating the Global Impact of DNA Methylation on Alternative Splicing. Cell Rep. 2015, 10, 1122–1134. [Google Scholar] [CrossRef]
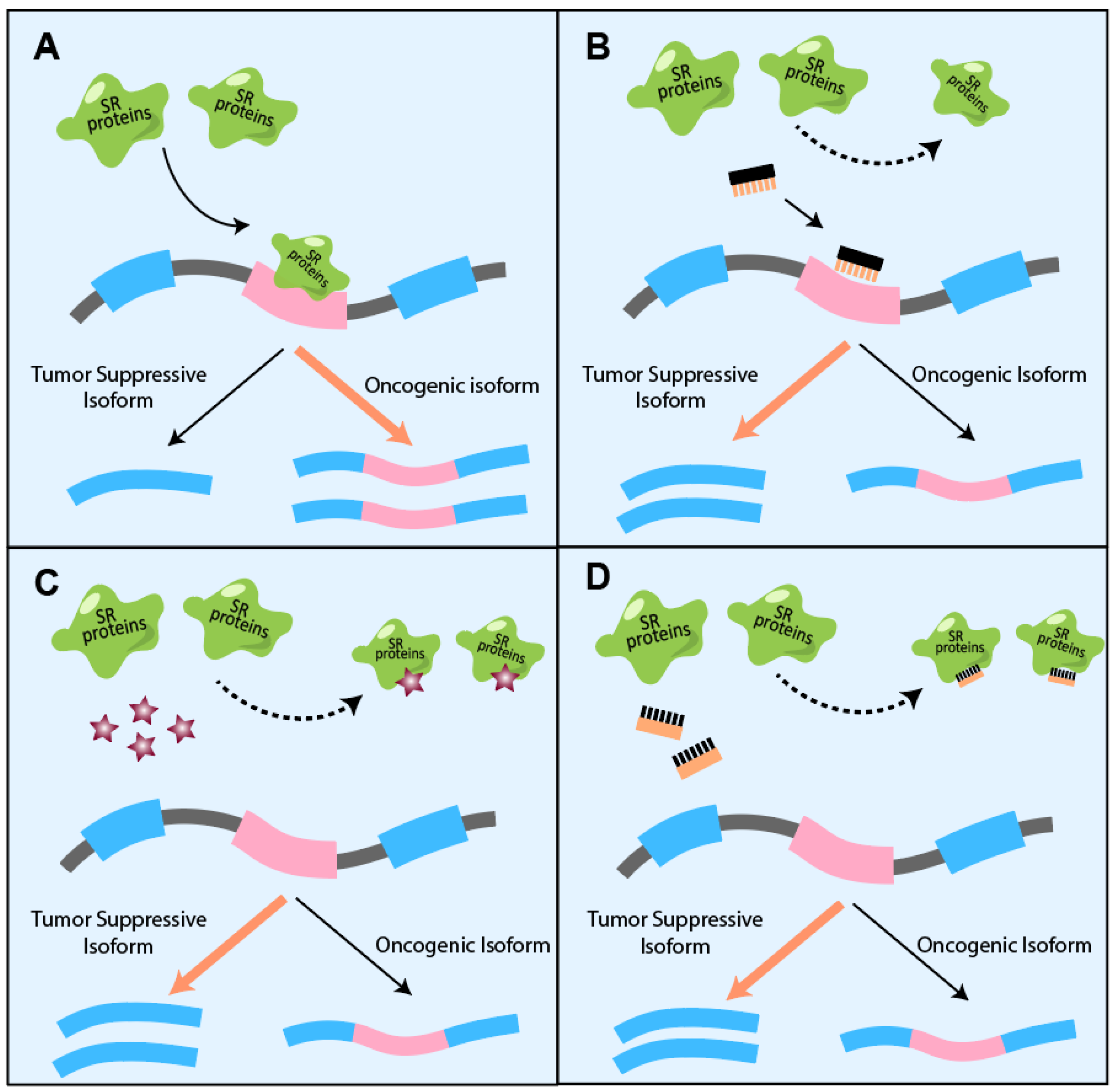
- Pandit, S.; Zhou, Y.; Shiue, L.; Coutinho-Mansfield, G.; Li, H.; Qiu, J.; Huang, J.; Yeo, G.W.; Ares, M.; Fu, X.-D. Genome-wide Analysis Reveals SR Protein Cooperation and Competition in Regulated Splicing. Mol. Cell 2013, 50, 223–235. [Google Scholar] [CrossRef]
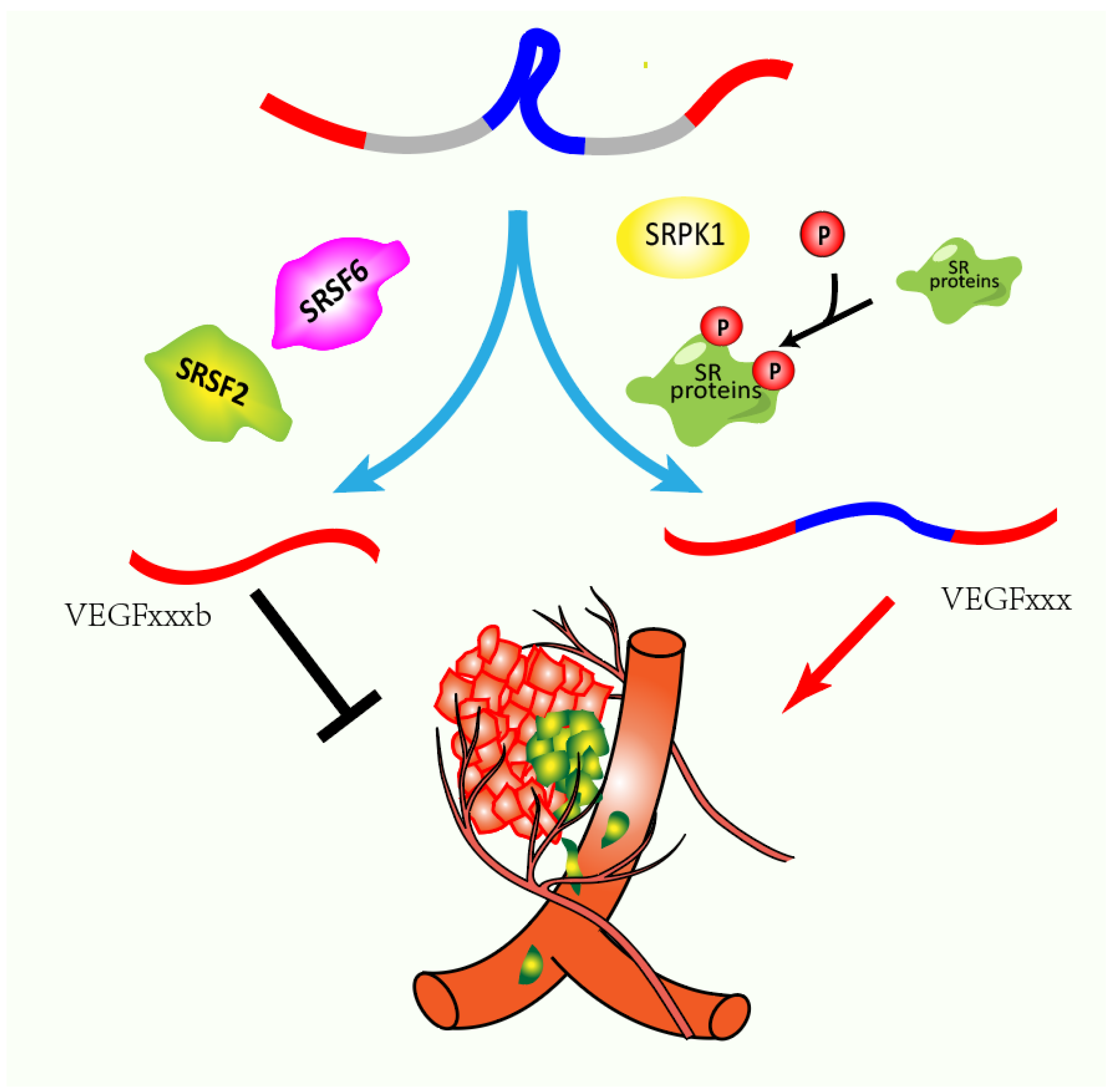
- Ourradi, K.; Blythe, T.; Jarrett, C.; Barratt, S.L.; Welsh, G.I.; Millar, A.B. VEGF isoforms have differential effects on permeability of human pulmonary microvascular endothelial cells. Respir. Res. 2017, 18, 116. [Google Scholar] [CrossRef]
- Perrin, R.M.; Konopatskaya, O.; Qiu, Y.; Harper, S.; Bates, D.O.; Churchill, A.J. Diabetic retinopathy is associated with a switch in splicing from anti- to pro-angiogenic isoforms of vascular endothelial growth factor. Diabetologia 2005, 48, 2422–2427. [Google Scholar] [CrossRef]
- Woolard, J.; Wang, W.-Y.; Bevan, H.S.; Qiu, Y.; Morbidelli, L.; Pritchard-Jones, R.O.; Cui, T.-G.; Sugiono, M.; Waine, E.; Perrin, R.; et al. VEGF165b, an Inhibitory Vascular Endothelial Growth Factor Splice Variant: Mechanism of Action, In vivo Effect On Angiogenesis and Endogenous Protein Expression. Cancer Res. 2004, 64, 7822–7835. [Google Scholar] [CrossRef]
- Pritchard-Jones, R.O.; Dunn, D.B.A.; Qiu, Y.; Varey, A.H.R.; Orlando, A.; Rigby, H.; Harper, S.J.; Bates, D.O. Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma. Br. J. Cancer 2007, 97, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz, R.; Peña, C.; Silva, J.; Lorenzo, Y.; García, V.; García, J.M.; Sánchez, A.; Espinosa, P.; Yuste, R.; Bonilla, F.; et al. p73 isoforms affect VEGF, VEGF165b and PEDF expression in human colorectal tumors: VEGF165b downregulation as a marker of poor prognosis. Int. J. Cancer 2008, 123, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Merdzhanova, G.; Gout, S.; Keramidas, M.; Edmond, V.; Coll, J.L.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. The transcription factor E2F1 and the SR protein SC35 control the ratio of pro-angiogenic versus antiangiogenic isoforms of vascular endothelial growth factor-A to inhibit neovascularization in vivo. Oncogene 2010, 29, 5392–5403. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Guiducci, S.; Romano, E.; Ceccarelli, C.; Bellando-Randone, S.; Conforti, M.L.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Overexpression of VEGF165b, an Inhibitory Splice Variant of Vascular Endothelial Growth Factor, Leads to Insufficient Angiogenesis in Patients With Systemic Sclerosis. Ital. J. Anat. Embryol. 2011, 109, e14–e26. [Google Scholar]
- Guyot, M.; Hilmi, C.; Ambrosetti, D.; Merlano, M.; Lo Nigro, C.; Durivault, J.; Grépin, R.; Pagès, G. Targeting the pro-angiogenic forms of VEGF or inhibiting their expression as anti-cancer strategies. Oncotarget 2016, 8, 9174. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Wang, Z.; Chatterjee, D.; Jeon, H.Y.; Akerman, M.; Vander Heiden, M.G.; Cantley, L.C.; Krainer, A.R. Exon-centric regulation of pyruvate kinase M alternative splicing via mutually exclusive exons. J. Mol. Cell Biol. 2011, 4, 79–87. [Google Scholar] [CrossRef]
- Kuranaga, Y.; Sugito, N.; Shinohara, H.; Tsujino, T.; Taniguchi, K.; Komura, K.; Ito, Y.; Soga, T.; Akao, Y. SRSF3, a Splicer of the PKM Gene, Regulates Cell Growth and Maintenance of Cancer-Specific Energy Metabolism in Colon Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3012. [Google Scholar] [CrossRef]
- Yadav, S.; Pant, D.; Samaiya, A.; Kalra, N.; Gupta, S.; Shukla, S. ERK1/2-EGR1-SRSF10 Axis Mediated Alternative Splicing Plays a Critical Role in Head and Neck Cancer. Front. Cell Dev. Biol. 2021, 9, 2468. [Google Scholar] [CrossRef]
- Ma, W.K.; Voss, D.M.; Scharner, J.; Costa, A.S.H.; Lin, K.-T.; Jeon, H.Y.; Wilkinson, J.E.; Jackson, M.; Rigo, F.; Bennett, C.F.; et al. ASO-Based PKM Splice-Switching Therapy Inhibits Hepatocellular Carcinoma Growth. Cancer Res. 2022, 82, 900–915. [Google Scholar] [CrossRef]
- Fukuhara, T.; Hosoya, T.; Shimizu, S.; Sumi, K.; Oshiro, T.; Yoshinaka, Y.; Suzuki, M.; Yamamoto, N.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc. Natl. Acad. Sci. USA 2006, 103, 11329–11333. [Google Scholar] [CrossRef]
- Pilch, B.; Allemand, E.; Facompré, M.; Bailly, C.; Riou, J.-F.; Soret, J.; Tazi, J. Specific Inhibition of Serine- and Arginine-rich Splicing Factors Phosphorylation, Spliceosome Assembly, and Splicing by the Antitumor Drug NB-506. Cancer Res. 2001, 61, 6876–6884. [Google Scholar]
- Bakkour, N.; Lin, Y.-L.; Maire, S.; Ayadi, L.; Mahuteau-Betzer, F.; Nguyen, C.H.; Mettling, C.; Portales, P.; Grierson, D.; Chabot, B.; et al. Small-Molecule Inhibition of HIV pre-mRNA Splicing as a Novel Antiretroviral Therapy to Overcome Drug Resistance. PLoS Pathog. 2007, 3, e159. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Li, J.; Du, X.; Zhang, W.; Lei, P.; Zhang, Q. Chimeric antibody targeting SRPK-1 in the treatment of non-small cell lung cancer by inhibiting growth, migration and invasion. Mol. Med. Rep. 2017, 16, 2121–2127. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, M.; Urtasun, R.; Elizalde, M.; Azkona, M.; Latasa, M.U.; Uriarte, I.; Arechederra, M.; Alignani, D.; Bárcena-Varela, M.; Álvarez-Sola, G.; et al. Splicing events in the control of genome integrity: Role of SLU7 and truncated SRSF3 proteins. Nucleic Acids Res. 2019, 47, 3450–3466. [Google Scholar] [CrossRef] [PubMed]
- Savisaar, R.; Hurst, L.D. Purifying Selection on Exonic Splice Enhancers in Intronless Genes. Mol. Biol. Evol. 2016, 33, 1396–1418. [Google Scholar] [CrossRef] [PubMed]
- Michlewski, G.; Sanford, J.R.; Cáceres, J.F. The Splicing Factor SF2/ASF Regulates Translation Initiation by Enhancing Phosphorylation of 4E-BP1. Mol. Cell 2008, 30, 179–189. [Google Scholar] [CrossRef]
- Denichenko, P.; Mogilevsky, M.; Cléry, A.; Welte, T.; Biran, J.; Shimshon, O.; Barnabas, G.D.; Danan-Gotthold, M.; Kumar, S.; Yavin, E.; et al. Specific inhibition of splicing factor activity by decoy RNA oligonucleotides. Nat. Commun. 2019, 10, 1590. [Google Scholar] [CrossRef]
- Shepard, P.J.; Hertel, K.J. The SR protein family. Genome Biol. 2009, 10, 242. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Splicing Factor Name | Cancer Type | Changes of SR Proteins in Cancer | SR Proteins’ Effect |
|---|---|---|---|
| SRSF1 | Lung Cancer | Protein [55,56] Phosphorylation [57] | Radioresistance [55] Autophagy [56] Apoptosis [57] |
| Breast Cancer | Protein [53] mRNA [49] | Apoptosis [49,53] Cell cycle arrest [49] | |
| Colon Cancer | mRNA [58] | DNA Damage [58] | |
| Glioma | mRNA and Protein [59] | Cytoskeleton reorganization [59] | |
| Renal Cancer | mRNA [60] | Apoptosis [60] | |
| SRSF2 | Lung Cancer | mRNA [61] | Angiopoiesis [61] |
| Liver Cancer | mRNA and Protein [62] | Proliferation [62] | |
| Renal Cancer | mRNA [60] | Apoptosis [60] | |
| SRSF3 | Ovarian Cancer | mRNA [63] | Apoptosis [63] |
| Colon Cancer | Protein [64] | Angiogenesis [64] | |
| Oral Cancer | Protein [65] | Autophagy [65] | |
| Glioma | mRNA [66] Protein [66] | Cell Mitosis [66] | |
| SRSF4 | Acute myeloid leukemia | mRNA [50] | Apoptosis [50] |
| SRSF5 | Pancreatic Cancer Breast Cancer | Phosphorylation [67] Protein [68] | Cell Cycle [67] Apoptosis [68] |
| SRSF6 | Colon Cancer Lung Cancer | mRNA [69] Protein [48] | Tumorigenesis [69] Apoptosis [70] Cell–cell junction [48] |
| SRSF7 | Colon Cancer Lung Cancer | Protein [71] mRNA [72] | Apoptosis [71] Growth Arrest [72] |
| SRSF9 | Colon Cancer | mRNA [73] Protein [73] | Ferroptosis [74] m6A Modification [73] |
| SRSF10 | Cervical Cancer | mRNA [75,76] Protein [75] | Macrophage Phagocytosis [75] Nonsense-mediated mRNA decay [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, L.; Deng, M.; Zhang, H. SR Splicing Factors Promote Cancer via Multiple Regulatory Mechanisms. Genes 2022, 13, 1659. https://doi.org/10.3390/genes13091659
Wan L, Deng M, Zhang H. SR Splicing Factors Promote Cancer via Multiple Regulatory Mechanisms. Genes. 2022; 13(9):1659. https://doi.org/10.3390/genes13091659
Chicago/Turabian StyleWan, Ledong, Min Deng, and Honghe Zhang. 2022. "SR Splicing Factors Promote Cancer via Multiple Regulatory Mechanisms" Genes 13, no. 9: 1659. https://doi.org/10.3390/genes13091659