
TBX3 and EFNA4 Variant in a Family with Ulnar-Mammary Syndrome and Sagittal Craniosynostosis
 , , , and
, , , and 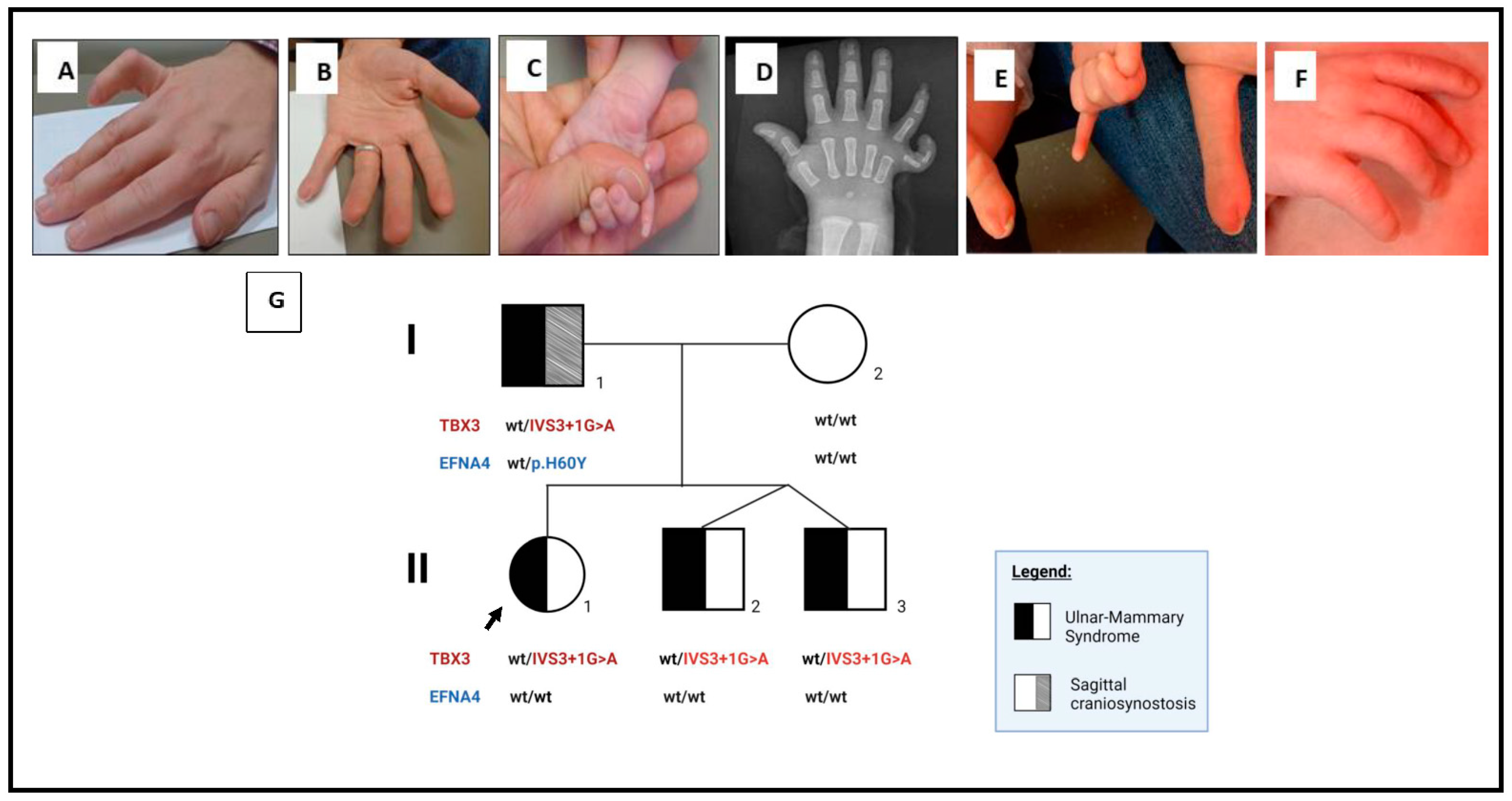{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Detailed Case Description
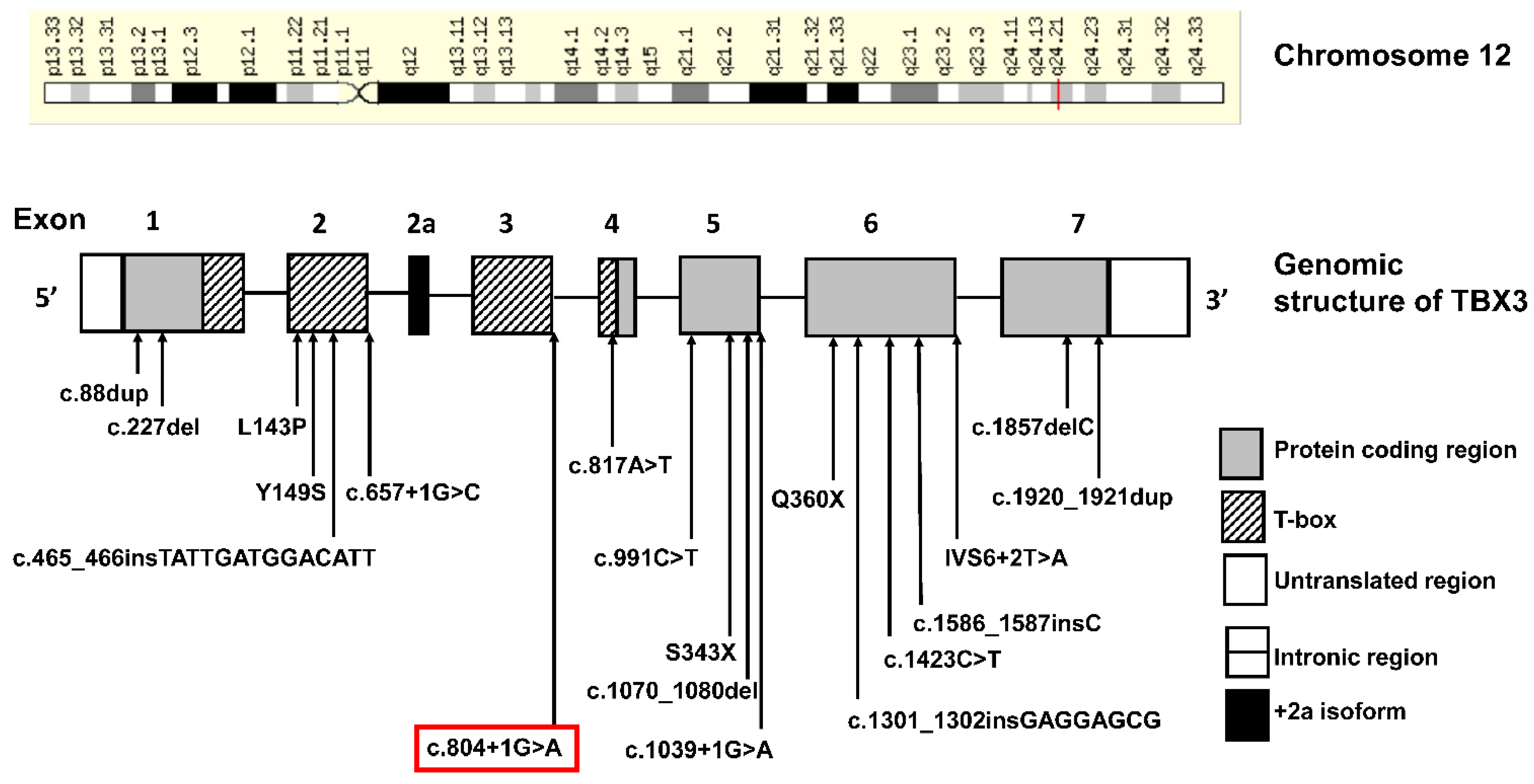
3. Genetics Testing
Materials and Methods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Qattan, M.M.; Maddirevula, S.; Alkuraya, F.S. A de novo TBX3 mutation presenting as dorsalization of the little fingers: A forme fruste phenotype of ulnar-mammary syndrome. Eur. J. Med. Genet. 2020, 63, 103615. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.; Le, T.; Watkins, W.; Dixon, M.; Kramer, B.; Roeder, A.; Carey, J.; Root, S.; Schinzel, A.; Van Maldergem, L.; et al. The Spectrum of Mutations in TBX3: Genotype/Phenotype Relationship in Ulnar-Mammary Syndrome. Am. J. Hum. Genet. 1999, 64, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, G.; Ogata, T.; Ishii, T.; Hasegawa, T.; Sato, S.; Matsuo, N. Novel mutation ofTBX3 in a Japanese family with Ulnar-Mammary syndrome: Implication for impaired sex development. Am. J. Med Genet. 2002, 110, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Linden, H.; Williams, R.; King, J.; Blair, E.; Kini, U. Ulnar mammary syndrome and TBX3: Expanding the phenotype. Am. J. Med Genet. Part A 2009, 149A, 2809–2812. [Google Scholar] [CrossRef]
- Meneghini, V.; Odent, S.; Platonova, N.; Egeo, A.; Merlo, G.R. Novel TBX3 mutation data in families with Ulnar–Mammary syndrome indicate a genotype–phenotype relationship: Mutations that do not disrupt the T-domain are associated with less severe limb defects. Eur. J. Med Genet. 2006, 49, 151–158. [Google Scholar] [CrossRef]
- Wollnik, B.; Kayserili, H.; Uyguner, O.; Tukel, T.; Yuksel-Apak, M. Haploinsufficiency of TBX3 causes ulnar-mammary syndrome in a large Turkish family. Ann Genet. 2002, 45, 213–217. [Google Scholar] [CrossRef]
- Klopocki, E.; Neumann, L.M.; Tonnies, H.; Ropers, H.H.; Mundlos, S.; Ullmann, R. Ulnar-Mammary Syndrome with Dysmorphic Facies and Mental Retardation Caused by a Novel 1.28 Mb Deletion Encompassing the Tbx3 Gene. Eur. J. Hum. Genet. 2006, 14, 1274–1279. [Google Scholar] [CrossRef]
- Carlson, H.; Ota, S.; Campbell, C.E.; Hurlin, P.J. A dominant repression domain in Tbx3 mediates transcriptional repression and cell immortalization: Relevance to mutations in Tbx3 that cause ulnar-mammary syndrome. Hum. Mol. Genet. 2001, 10, 2403–2413. [Google Scholar] [CrossRef]
- Frank, D.U.; Emechebe, U.; Thomas, K.R.; Moon, A.M. Mouse Tbx3 Mutants Suggest Novel Molecular Mechanisms for Ulnar-Mammary Syndrome. PLoS ONE 2013, 8, e67841. [Google Scholar] [CrossRef]
- Yilmaz, E.; Mihci, E.; Nur, B.; Alper, Ö.M.; Taçoy, Ş. Recent Advances in Craniosynostosis. Pediatr. Neurol. 2019, 99, 7–15. [Google Scholar] [CrossRef]
- Lattanzi, W.; Barba, M.; Di Pietro, L.; Boyadjiev, S.A. Genetic Advances in Craniosynostosis. Am. J. Med. Genet. Part A 2017, 173, 1406–1429. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, A.O.; Johnson, D.; Wall, S.A. Clinical genetics of craniosynostosis. Curr. Opin. Pediatr. 2017, 29, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Justice, C.M.; Yagnik, G.; Kim, Y.; Peter, I.; Wang Jabs, E.; Erazo, M.; Ye, X.; Ainehsazan, E.; Shi, L.; Cunningham, M.L.; et al. A Genome-Wide Association Study Identifies Susceptibility Loci for Nonsyndromic Sagittal Craniosynostosis near Bmp2 and within Bbs9. Nat. Genet. 2012, 44, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, V.E. The T-Box Gene Family: Emerging Roles in Development, Stem Cells and Cancer. Development 2014, 141, 3819–3833. [Google Scholar] [CrossRef]
- Packham, E.A.; Brook, J.D. T-box genes in human disorders. Hum. Mol. Genet. 2003, 12, R37–R44. [Google Scholar] [CrossRef]
- Bamshad, M.; Krakowiak, P.A.; Watkins, W.S.; Root, S.; Carey, J.C.; Jorde, L.B. A Gene for Ulnar-Mammary Syndrome Maps to 12q23-Q24.1. Hum. Mol. Genet. 1995, 4, 1973–1977. [Google Scholar] [CrossRef]
- Bamshad, M.; Lin, R.C.; Law, D.J.; Watkins, W.S.; Krakowiak, P.A.; Moore, M.E.; Franceschini, P.; Lala, R.; Holmes, L.B.; Gebuhr, T.C.; et al. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat. Genet. 1997, 16, 311–315. [Google Scholar] [CrossRef]
- Khan, S.F.; Damerell, V.; Omar, R.; Du Toit, M.; Khan, M.; Maranyane, H.M.; Mlaza, M.; Bleloch, J.; Bellis, C.; Sahm, B.D.; et al. The roles and regulation of TBX3 in development and disease. Gene 2020, 726, 144223. [Google Scholar] [CrossRef]
- Willmer, T.; Cooper, A.; Peres, J.; Omar, R.; Prince, S. The T-Box transcription factor 3 in development and cancer. Biosci. Trends 2017, 11, 254–266. [Google Scholar] [CrossRef]
- Tanteles, G.; Nicolaou, N.; Syrimis, A.; Metaxa, R.; Nicolaou, M.; Christophidou-Anastasiadou, V.; Skordis, N. Novel TBX3 mutation in a family of Cypriot ancestry with ulnar-mammary syndrome. Clin. Dysmorphol. 2017, 26, 61–65. [Google Scholar] [CrossRef]
- Kumar, P.P.; Franklin, S.; Emechebe, U.; Hu, H.; Moore, B.; Lehman, C.; Yandell, M.; Moon, A.M. TBX3 Regulates Splicing In Vivo: A Novel Molecular Mechanism for Ulnar-Mammary Syndrome. PLoS Genet. 2014, 10, e1004247. [Google Scholar] [CrossRef] [PubMed]
- Merrill, A.E.; Bochukova, E.G.; Brugger, S.M.; Ishii, M.; Pilz, D.T.; Wall, S.A.; Lyons, K.M.; Wilkie, A.O.; Maxson, R.E. Cell mixing at a neural crest-mesoderm boundary and deficient ephrin-Eph signaling in the pathogenesis of craniosynostosis. Hum. Mol. Genet. 2006, 15, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.M.; Fok, V.T.; Gustafson, J.A.; Smyth, M.D.; Timms, A.E.; Frazar, C.D.; Smith, J.D.; Birgfeld, C.B.; Lee, A.; Ellenbogen, R.G.; et al. Single suture craniosynostosis: Identification of rare variants in genes associated with syndromic forms. Am. J. Med. Genet. Part A 2018, 176, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Fonteles, C.; Finnell, R.; Lei, Y.; Zurita-Jimenez, M.; Monteiro, A.; George, T.; Harshbarger, R. De novo ALX4 variant detected in child with non-syndromic craniosynostosis. Braz. J. Med. Biol. Res. 2021, 54, e11396. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tung, M.L.; Chandra, B.; Kotlarek, J.; Melo, M.; Phillippi, E.; Justice, C.M.; Musolf, A.; Boyadijev, S.A.; Romitti, P.A.; Darbro, B.; et al. TBX3 and EFNA4 Variant in a Family with Ulnar-Mammary Syndrome and Sagittal Craniosynostosis. Genes 2022, 13, 1649. https://doi.org/10.3390/genes13091649
Tung ML, Chandra B, Kotlarek J, Melo M, Phillippi E, Justice CM, Musolf A, Boyadijev SA, Romitti PA, Darbro B, et al. TBX3 and EFNA4 Variant in a Family with Ulnar-Mammary Syndrome and Sagittal Craniosynostosis. Genes. 2022; 13(9):1649. https://doi.org/10.3390/genes13091649
Chicago/Turabian StyleTung, Moon Ley, Bharatendu Chandra, Jaclyn Kotlarek, Marcelo Melo, Elizabeth Phillippi, Cristina M. Justice, Anthony Musolf, Simeon A. Boyadijev, Paul A. Romitti, Benjamin Darbro, and et al. 2022. "TBX3 and EFNA4 Variant in a Family with Ulnar-Mammary Syndrome and Sagittal Craniosynostosis" Genes 13, no. 9: 1649. https://doi.org/10.3390/genes13091649