
Evaluation of Intracellular Gene Transfers from Plastome to Nuclear Genome across Progressively Improved Assemblies for Arabidopsis thaliana and Oryza sativa

Abstract
:1. Introduction
2. Methods
2.1. Data Sampling
2.1.1. Acquisition of Genomic Data
2.1.2. Quality Assessment
2.2. Analysis of IGT
Sequence Alignment
2.3. Plastomes Transferred to Nuclear Chromosomes
2.4. Plastomes Transferred to Nuclear Genomic Regions
2.5. Characteristics of the Transferred Fragments
3. Results
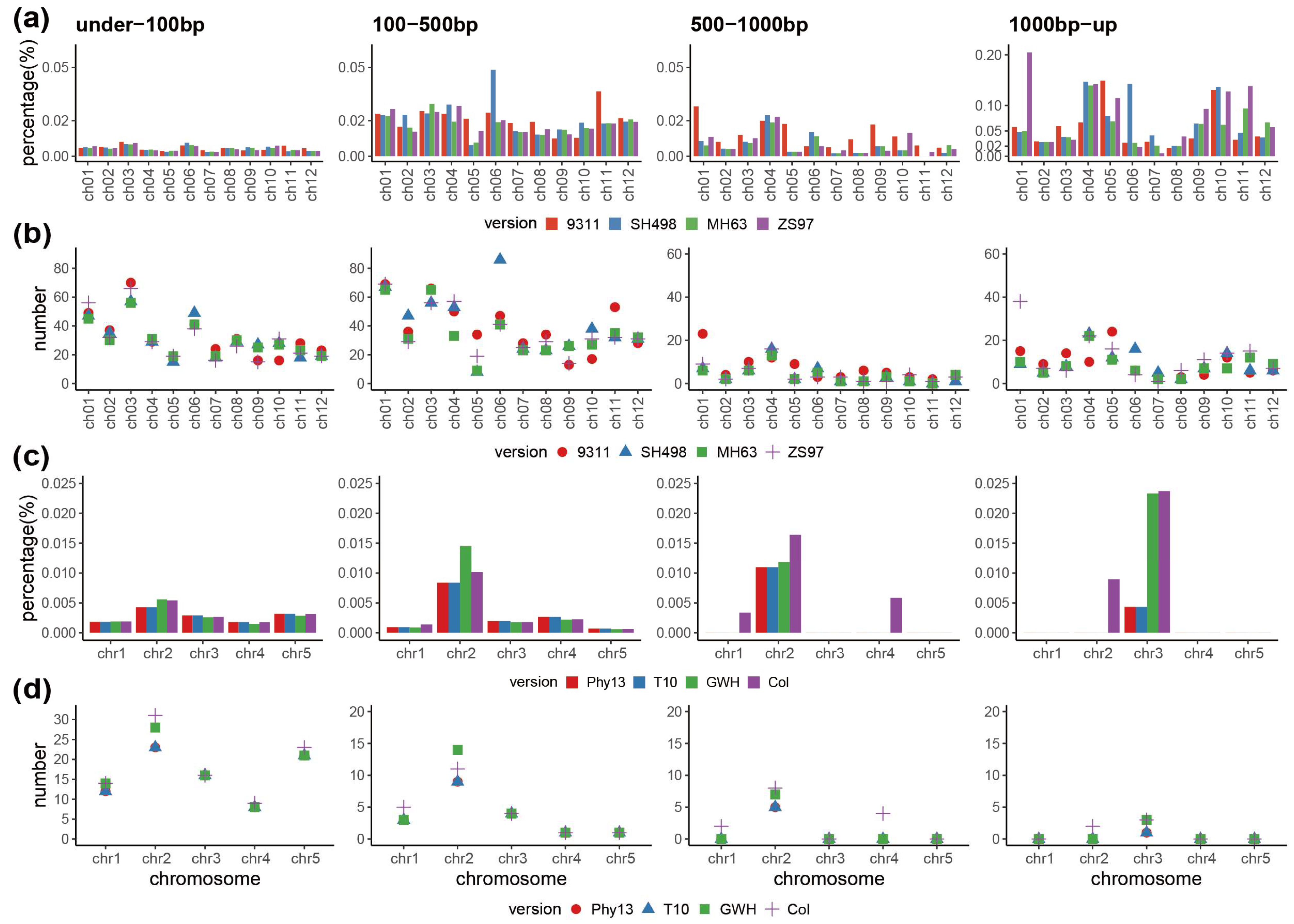
3.1. Patterns in IGT from Plastome to Nuclear Genome in A. thaliana and O. sativa
3.2. Characterization of the Transfer Fragments and Flanking Sequences
3.3. Differences of Plastome to Nuclear Genome IGT among Assembly Versions and Genomic Regions
4. Discussion
4.1. IGT Occurs Continuously and Can Be Detected More Accurately with Improved Genome Assembly
4.2. TEs May Be a Factor Involved in Mediating IGT
4.3. IGT as a Possible New Index to Assess Genome Assemblies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Altemose, N.; Logsdon, G.A.; Bzikadze, A.V.; Sidhwani, P.; Langley, S.A.; Caldas, G.V.; Hoyt, S.J.; Uralsky, L.; Ryabov, F.D.; Shew, C.J.; et al. Complete genomic and epigenetic maps of human centromeres. Science 2022, 376, eabl4178. [Google Scholar] [CrossRef] [PubMed]
- Naish, M.; Alonge, M.; Wlodzimierz, P.; Tock, A.J.; Abramson, B.W.; Schmücker, A.; Mandáková, T.; Jamge, B.; Lambing, C.; Kuo, P.; et al. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science 2021, 374, eabi7489. [Google Scholar] [CrossRef]
- Song, J.-M.; Xie, W.-Z.; Wang, S.; Guo, Y.-X.; Koo, D.-H.; Kudrna, D.; Gong, C.; Huang, Y.; Feng, J.-W.; Zhang, W.; et al. Two gap-free reference genomes and a global view of the centromere architecture in rice. Mol. Plant 2021, 14, 1757–1767. [Google Scholar] [CrossRef]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.-S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Avni, R.; Nave, M.; Barad, O.; Baruch, K.; Twardziok, S.O.; Gundlach, H.; Hale, I.; Mascher, M.; Spannagl, M.; Wiebe, K.; et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 2017, 357, 93–97. [Google Scholar] [CrossRef]
- Gui, S.; Peng, J.; Wang, X.; Wu, Z.; Cao, R.; Salse, J.; Zhang, H.; Zhu, Z.; Xia, Q.; Quan, Z.; et al. Improving Nelumbo nucifera genome assemblies using high-resolution genetic maps and BioNano genome mapping reveals ancient chromosome rearrangements. Plant J. 2018, 94, 721–734. [Google Scholar] [CrossRef]
- Jaillon, O.; Aury, J.-M.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [CrossRef] [Green Version]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Wang, B.; Yang, X.; Jia, Y.; Xu, Y.; Jia, P.; Dang, N.; Wang, S.; Xu, T.; Zhao, X.; Gao, S.; et al. High-quality Arabidopsis thaliana genome assembly with nanopore and HiFi long reads. Genom. Proteom. Bioinform. 2021, in press. [Google Scholar] [CrossRef]
- Ou, S.; Chen, J.; Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 2018, 46, e126. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Bergthorsson, U.; Adams, K.L.; Thomason, B.; Palmer, J.D. Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature 2003, 424, 197–201. [Google Scholar] [CrossRef]
- Filip, E.; Skuza, L. Horizontal gene transfer involving chloroplasts. Int. J. Mol. Sci. 2021, 22, 4484. [Google Scholar] [CrossRef]
- Bock, R. The give-and-take of DNA: Horizontal gene transfer in plants. Trends Plant Sci. 2010, 15, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.; Bhattacharya, D. Horizontal gene transfer in eukaryotes: Not if, but how much? Trends Genet. 2020, 36, 915–925. [Google Scholar] [CrossRef]
- Gao, C.; Ren, X.; Mason, A.S.; Liu, H.; Xiao, M.; Li, J.; Fu, D. Horizontal gene transfer in plants. Funct. Integr. Genom. 2014, 14, 23–29. [Google Scholar] [CrossRef]
- Timmis, J.N.; Ayliffe, M.A.; Huang, C.Y.; Martin, W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004, 5, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Handa, H. The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): Comparative analysis of the mitochondrial genomes of rapeseed and Arab. Thaliana Nucleic Acids Res. 2003, 31, 5907–5916. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, Y.; Yamazaki, Y.; Murai, K.; Kanno, A.; Terachi, T.; Shiina, T.; Miyashita, N.; Nasuda, S.; Nakamura, C.; Mori, N.; et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 2005, 33, 6235–6250. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, Y.; Watase, Y.; Nagase, M.; Makita, N.; Yagura, S.; Hirai, A.; Sugiura, M. The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: Comparative analysis of mitochondrial genomes in higher plants. Mol. Genet. Genom. 2005, 272, 603–615. [Google Scholar] [CrossRef]
- Shahmuradov, I.A.; Akbarova, Y.Y.; Solovyev, V.V.; Aliyev, J.A. Abundance of plastid DNA insertions in nuclear genomes of rice and Arabidopsis. Plant Mol. Biol. 2003, 52, 923–934. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, J.; Breen, J.; Kong, X. Recent insertion of a 52-kb mitochondrial DNA segment in the wheat lineage. Funct. Integr. Genom. 2011, 11, 599–609. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, T.; Wang, X.; Qu, J.; Jia, X. The complete chloroplast genome of Sargassum horneri and its phylogenetic analysis. Mitochondrial DNA Part B 2019, 4, 3312–3313. [Google Scholar] [CrossRef]
- Straub, S.C.K.; Cronn, R.C.; Edwards, C.; Fishbein, M.; Liston, A. Horizontal transfer of DNA from the mitochondrial to the plastid genome and its subsequent evolution in milkweeds (Apocynaceae). Genome Biol. Evol. 2013, 5, 1872–1885. [Google Scholar] [CrossRef]
- Smith, D.R. Extending the limited transfer window hypothesis to inter-organelle DNA migration. Genome Biol. Evol. 2011, 3, 743–748. [Google Scholar] [CrossRef]
- Leister, D. Origin, evolution and genetic effects of nuclear insertions of organelle DNA. Trends Genet. 2005, 21, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Kleine, T.; Maier, U.G.; Leister, D. DNA transfer from organelles to the nucleus: The idiosyncratic genetics of endosymbiosis. Annu. Rev. Plant Biol. 2009, 60, 115–138. [Google Scholar] [CrossRef]
- Choi, K.-S.; Park, S. Complete plastid and mitochondrial genomes of Aeginetia indica reveal intracellular gene transfer (IGT), horizontal gene transfer (HGT), and cytoplasmic male sterility (CMS). Int. J. Mol. Sci. 2021, 22, 6143. [Google Scholar] [CrossRef]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef]
- Zhao, N.; Wang, Y.; Hua, J. The roles of mitochondrion in intergenomic gene transfer in plants: A source and a pool. Int. J. Mol. Sci. 2018, 19, 547. [Google Scholar] [CrossRef]
- Du, H.; Yu, Y.; Ma, Y.; Gao, Q.; Cao, Y.; Chen, Z.; Ma, B.; Qi, M.; Li, Y.; Zhao, X.; et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 2017, 8, 15324. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Hu, G.; Kurgan, L. Sequence similarity searching. Curr. Protoc. Protein Sci. 2018, 95, e71. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Ou, S.; Hufford, M.B.; Peterson, T. A tutorial of EDTA: Extensive de novo TE annotator. In Plant Transposable Elements: Methods in Molecular Biology; Cho, J., Ed.; Springer: New York, NY, USA, 2021; pp. 55–67. [Google Scholar]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Fields, P.D.; Waneka, G.; Naish, M.; Schatz, M.C.; Henderson, I.R.; Sloan, D.B. Complete sequence of a 641-kb insertion of mitochondrial DNA in the Arabidopsis thaliana nuclear genome. Genome Biol. Evol. 2022, 14, evac059. [Google Scholar] [CrossRef]
- Hazkani-Covo, E.; Zeller, R.M.; Martin, W. Molecular poltergeists: Mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 2010, 6, e1000834. [Google Scholar] [CrossRef]
- Zhang, G.-J.; Dong, R.; Lan, L.-N.; Li, S.-F.; Gao, W.-J.; Niu, H.-X. Nuclear integrants of organellar DNA contribute to genome structure and evolution in plants. Int. J. Mol. Sci. 2020, 21, 707. [Google Scholar] [CrossRef]
- Michalovova, M.; Vyskot, B.; Kejnovsky, E. Analysis of plastid and mitochondrial DNA insertions in the nucleus (NUPTs and NUMTs) of six plant species: Size, relative age and chromosomal localization. Heredity 2013, 111, 314–320. [Google Scholar] [CrossRef]
- Huang, C.Y.; Grünheit, N.; Ahmadinejad, N.; Timmis, J.N.; Martin, W. Mutational decay and age of chloroplast and mitochondrial genomes transferred recently to angiosperm nuclear chromosomes. Plant Physiology. 2005, 138, 1723–1733. [Google Scholar] [CrossRef]
- Cullis, C.A.; Vorster, B.J.; Van Der Vyver, C.; Kunert, K.J. Transfer of genetic material between the chloroplast and nucleus: How is it related to stress in plants? Ann. Bot. 2008, 103, 625–633. [Google Scholar] [CrossRef]
- Roark, L.M.; Hui, A.Y.; Donnelly, L.; Birchler, J.A.; Newton, K.J. Recent and frequent insertions of chloroplast DNA into maize nuclear chromosomes. Cytogenet. Genome Res. 2010, 129, 17–23. [Google Scholar] [CrossRef]
- Richly, E.; Leister, D. NUMTs in sequenced eukaryotic genomes. Mol. Biol. Evol. 2004, 21, 1081–1084. [Google Scholar] [CrossRef]
- Lin, X.; Kaul, S.; Rounsley, S.; Shea, T.P.; Benito, M.-I.; Town, C.D.; Fujii, C.Y.; Mason, T.; Bowman, C.L.; Barnstead, M.; et al. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature 1999, 402, 761–768. [Google Scholar] [CrossRef] [Green Version]
- Noutsos, C.; Richly, E.; Leister, D. Generation and evolutionary fate of insertions of organelle DNA in the nuclear genomes of flowering plants. Genome Res. 2005, 15, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, A.E.; Timmis, J.N. Instability of plastid DNA in the nuclear genome. PLoS Genet. 2009, 5, e1000323. [Google Scholar] [CrossRef] [PubMed]
- Bock, R.; Timmis, J.N. Reconstructing evolution: Gene transfer from plastids to the nucleus. Bioessays 2008, 30, 556–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Version | Name | Time | Ecotype | Assembly | Sequencing Tech | BUSCO |
|---|---|---|---|---|---|---|
| phytozome13 | Phy13 | 2013 | Columbia | Athaliana_167.fa.gz | Next-generation sequencing | 99.30% |
| tair10 | T10 | 2014 | Columbia | GCA_000001735.2 | Next-generation sequencing | 99.30% |
| almost complete | GWH | 2021 | Columbia | GWHBDNP00000000.1 | ONT and PacBio and Hi-C | 99.40% |
| no-gap | Col | 2021 | Columbia | Col-CEN | ONT and PacBio HiFi long-read | 99.40% |
| Version | Name | Time | Strain | Assembly | Sequencing Tech | BUSCO |
|---|---|---|---|---|---|---|
| draft sequence | 9311 | 2002 | Indica | GCA_000004675.2 | Whole-genome shotgun sequencing | 96.30% |
| near complete | SH498 | 2017 | Indica | GCA_002151415.1 | PacBio | 98.50% |
| no-gap | MH63 | 2021 | Indica | GCA_001623365.2 | PacBio Sequel II | 98.70% |
| no-gap | ZS97 | 2021 | Indica | GCA_001623345.3 | PacBio Sequel II | 98.70% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Liao, X.; Tembrock, L.R.; Yang, Z.; Wu, Z. Evaluation of Intracellular Gene Transfers from Plastome to Nuclear Genome across Progressively Improved Assemblies for Arabidopsis thaliana and Oryza sativa. Genes 2022, 13, 1620. https://doi.org/10.3390/genes13091620
Wang H, Liao X, Tembrock LR, Yang Z, Wu Z. Evaluation of Intracellular Gene Transfers from Plastome to Nuclear Genome across Progressively Improved Assemblies for Arabidopsis thaliana and Oryza sativa. Genes. 2022; 13(9):1620. https://doi.org/10.3390/genes13091620
Chicago/Turabian StyleWang, Haoqi, Xuezhu Liao, Luke R. Tembrock, Zuoren Yang, and Zhiqiang Wu. 2022. "Evaluation of Intracellular Gene Transfers from Plastome to Nuclear Genome across Progressively Improved Assemblies for Arabidopsis thaliana and Oryza sativa" Genes 13, no. 9: 1620. https://doi.org/10.3390/genes13091620