
Comparative Analysis of Complete Chloroplast Genomes of Nine Species of Litsea (Lauraceae): Hypervariable Regions, Positive Selection, and Phylogenetic Relationships
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, DNA Extraction, and Sequencing
2.2. Chloroplast Genome De Novo Assembly and Annotation
2.3. Analysis of Chloroplast Genome Characteristics
2.4. Comparative Analysis
2.5. Phylogenetic Analysis
3. Results and Discussion
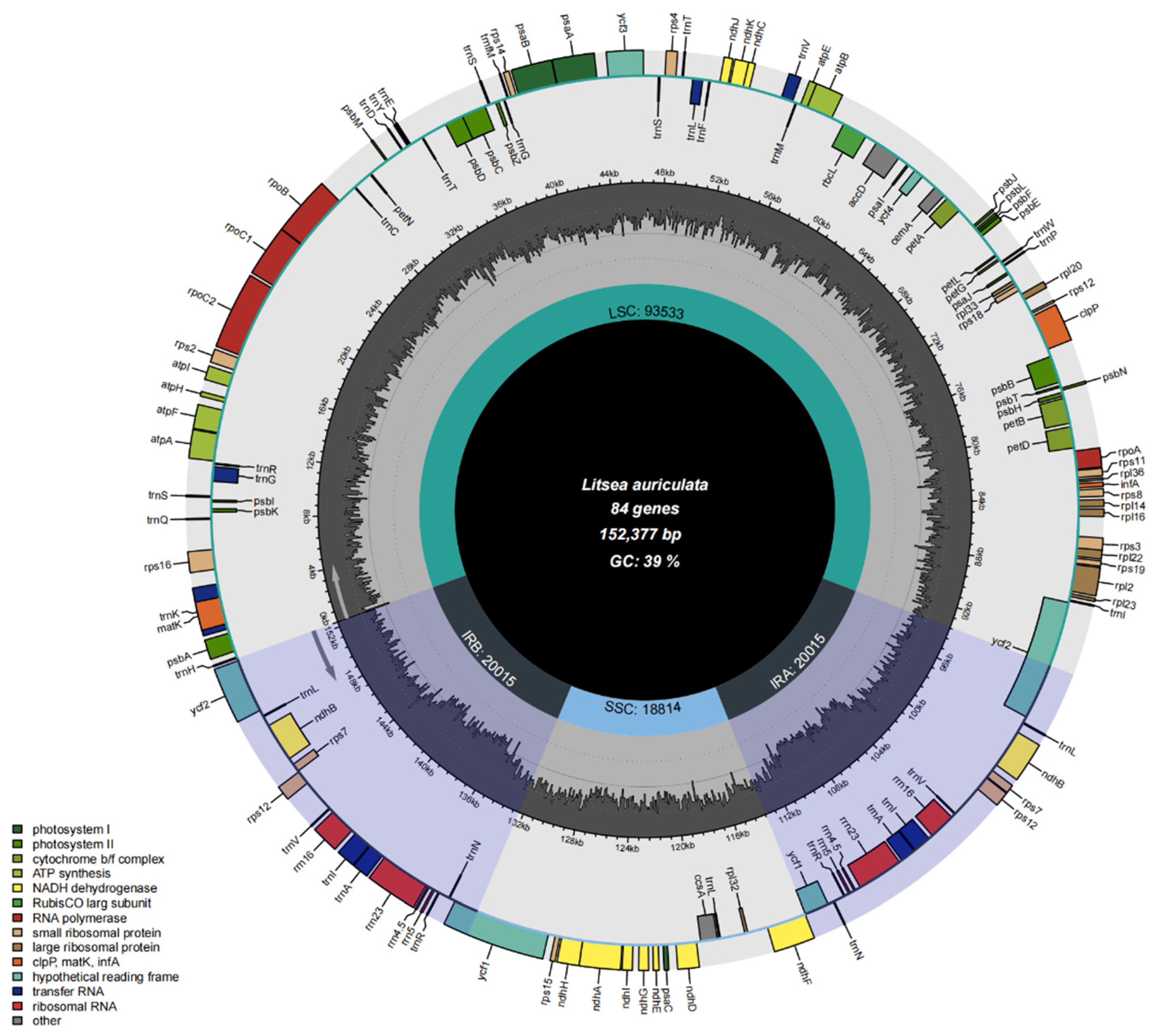
3.1. Chloroplast Genome Features of Litsea
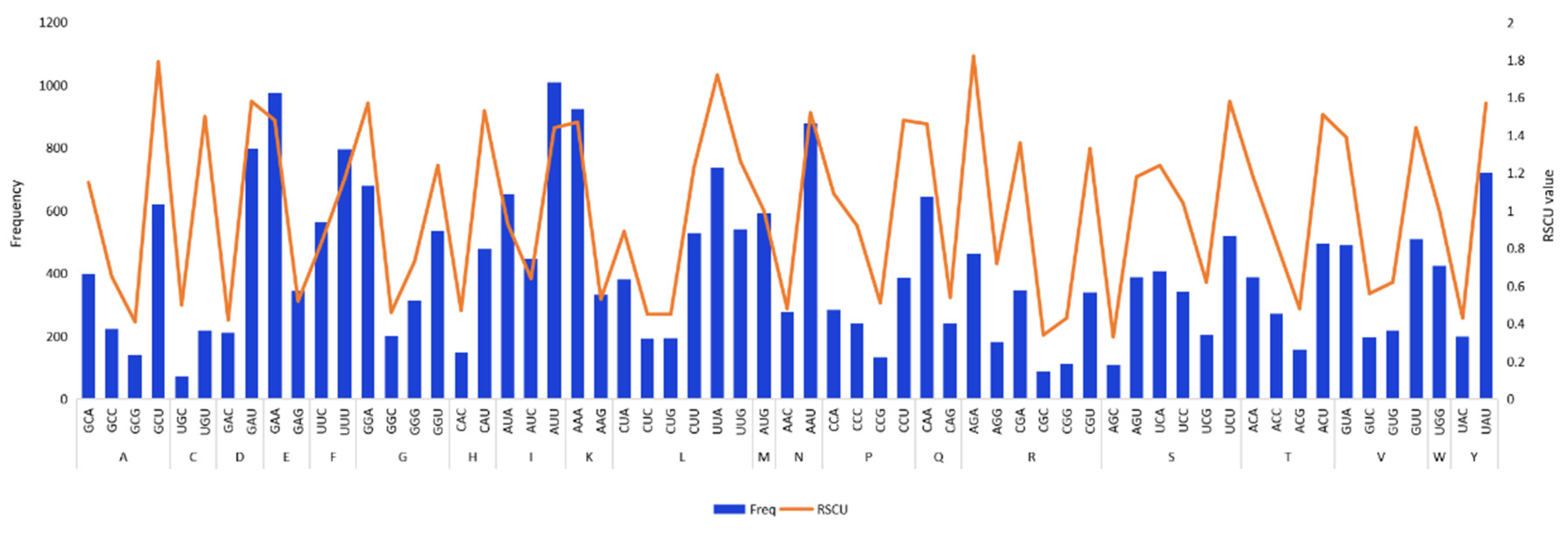
3.2. Codon Usage Analysis
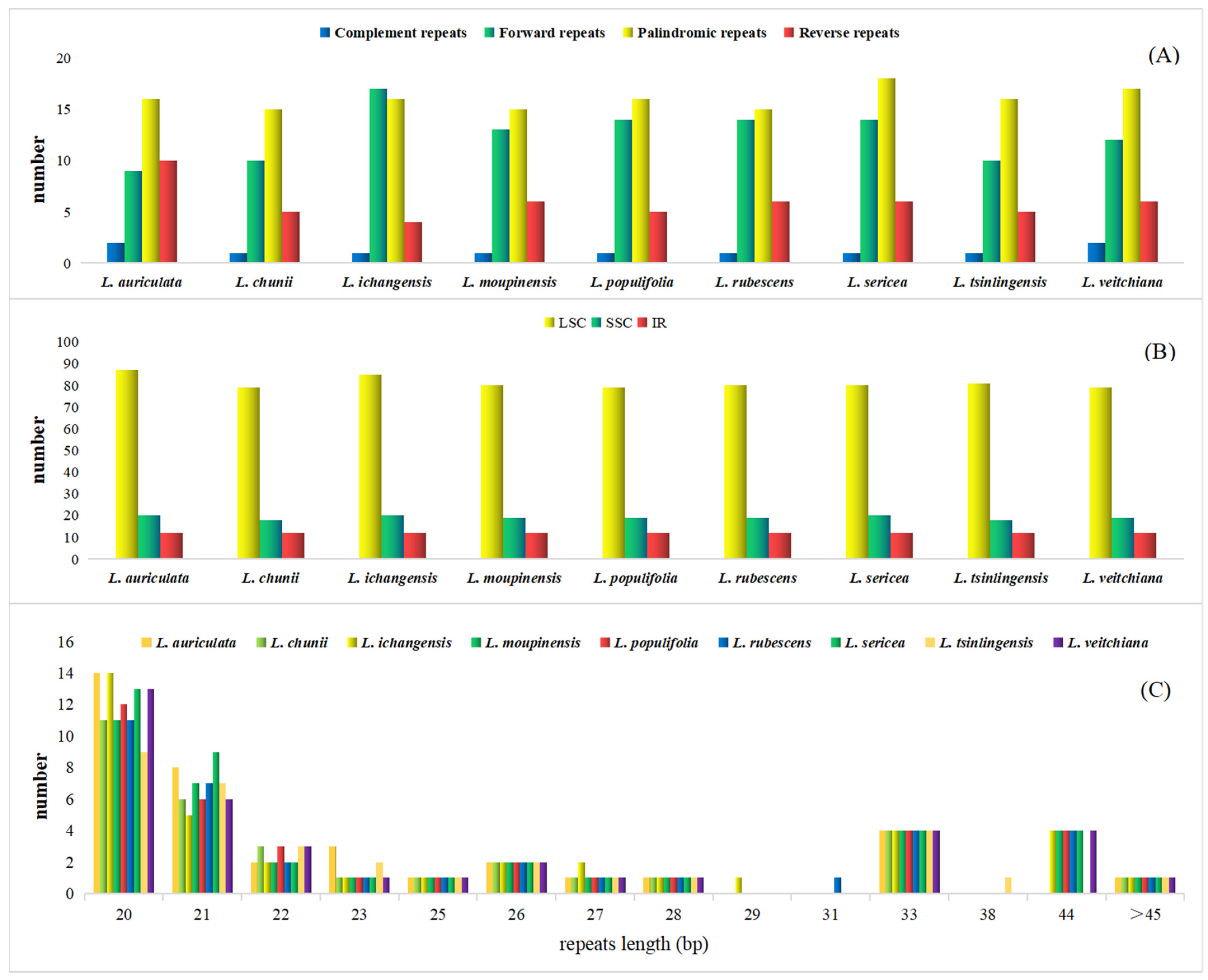
3.3. Long Repeat and SSR Analysis
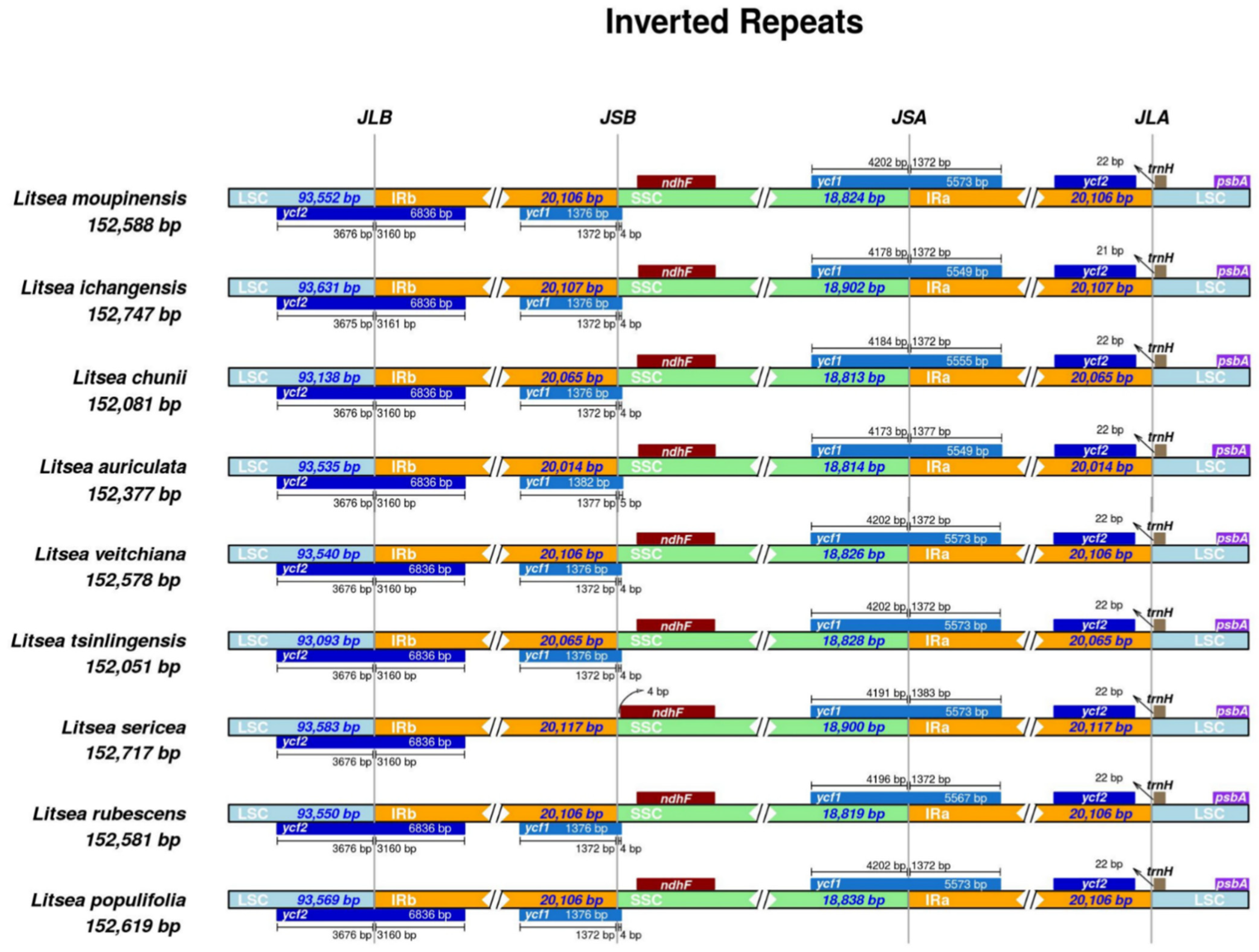
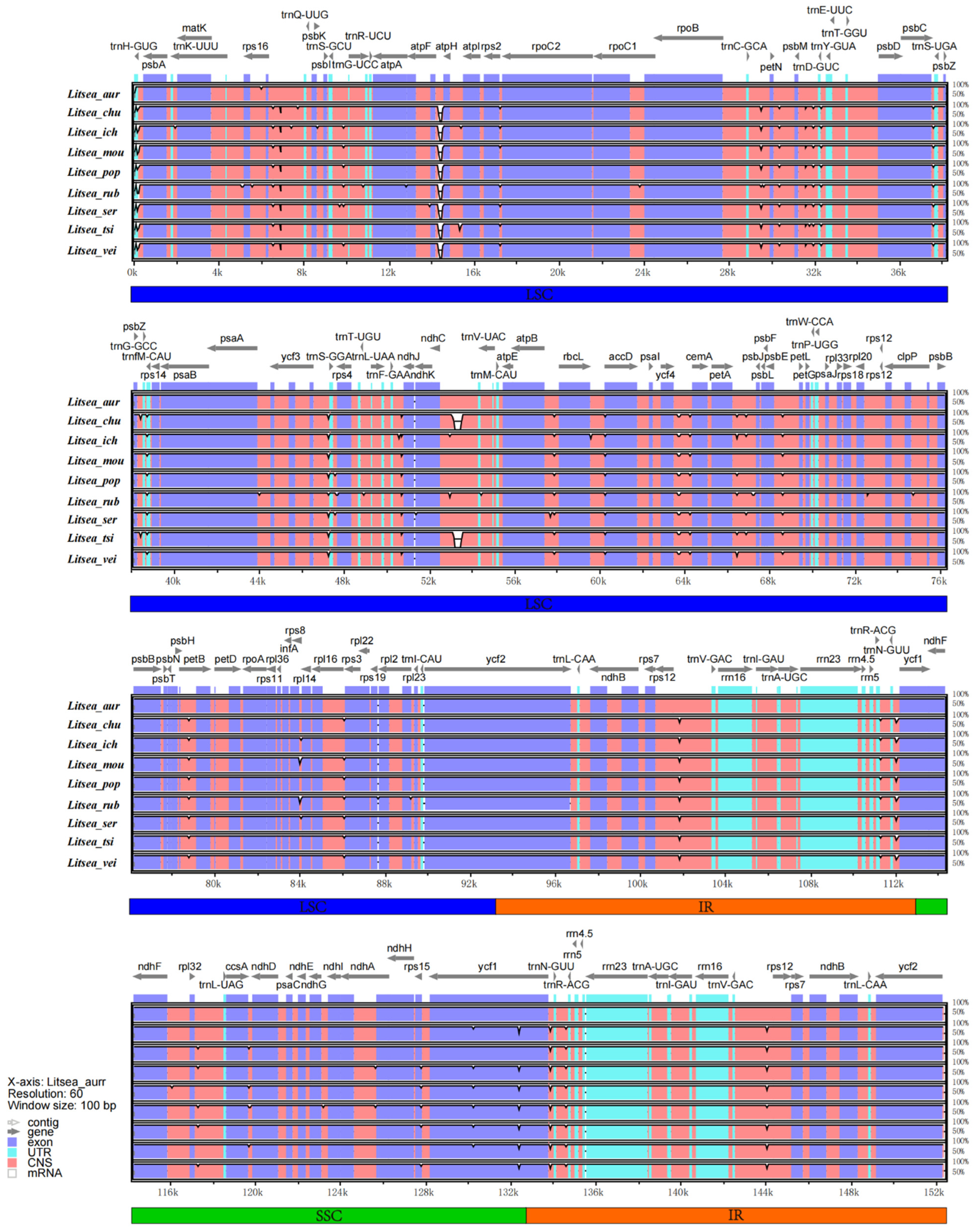
3.4. IR Contraction Analysis and Sequence Identity Plot
3.5. SNP and InDels
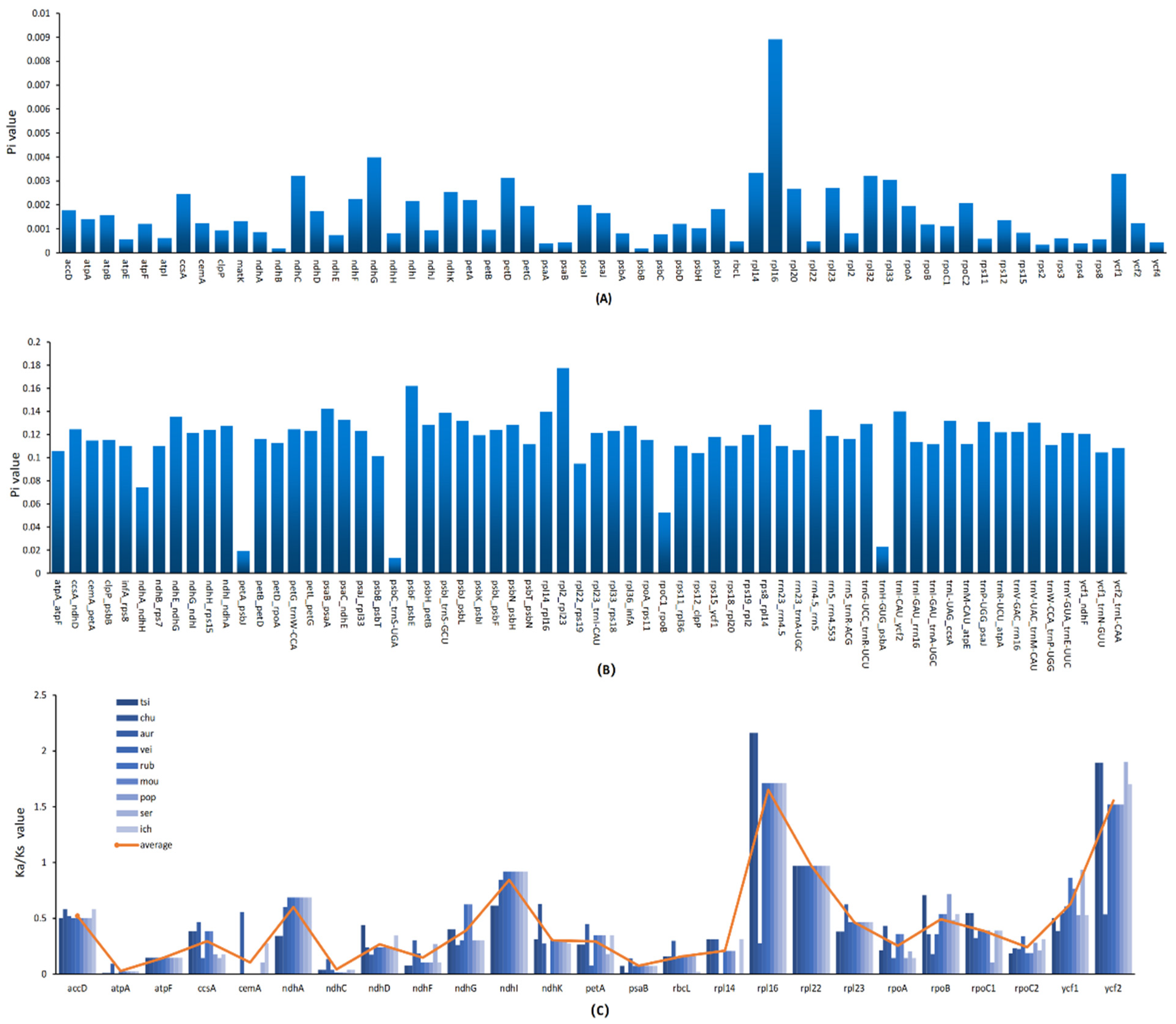
3.6. Nucleotide Divergence and Selection Pressure
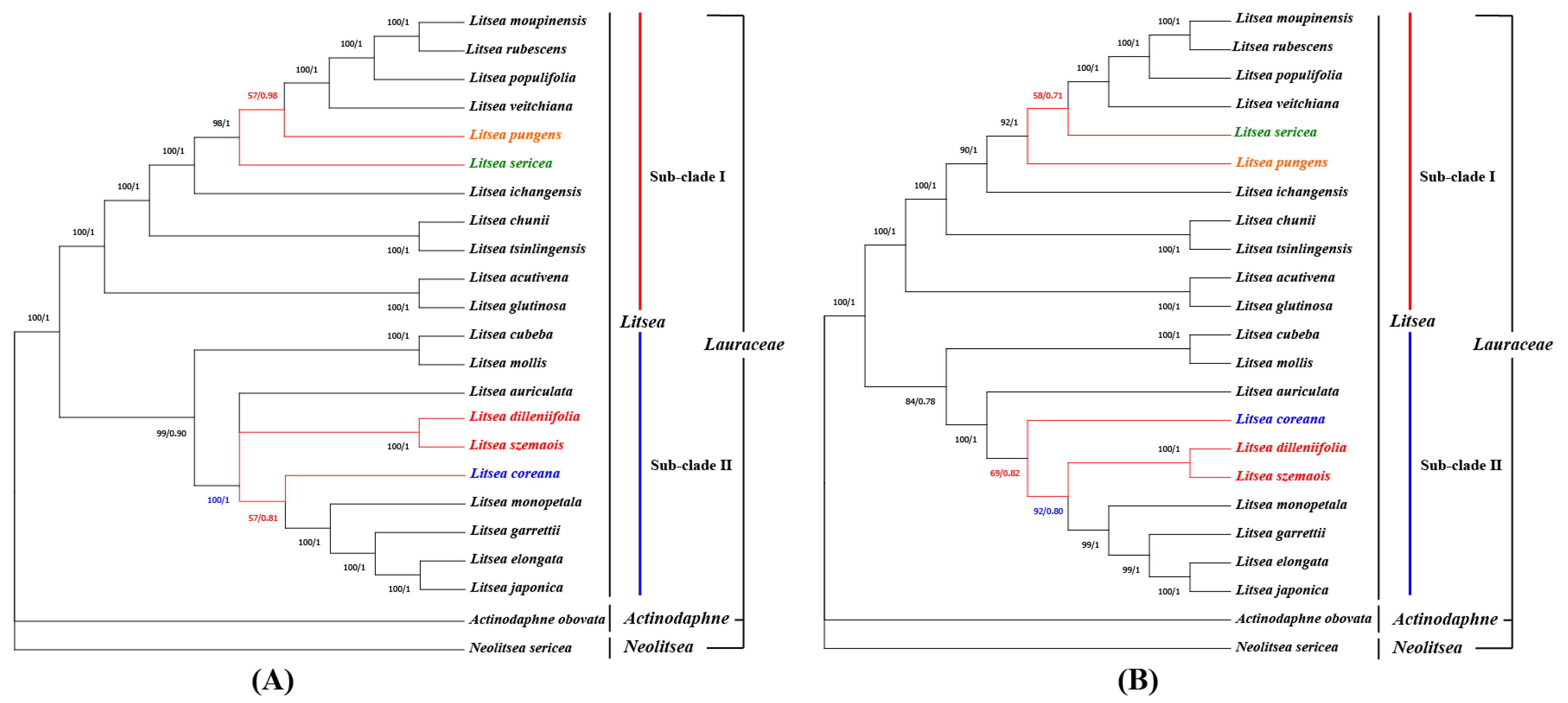
3.7. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Wang, Y.S.; Wen, Z.Q.; Li, B.T.; Zhang, H.B.; Yang, J.H. Ethnobotany, phytochemistry, and pharmacology of the genus Litsea: An update. J. Ethnopharmacol. 2016, 181, 66–107. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.G.; Zhao, Y.; Li, G.H.; Chen, B.J.; Wang, X.N.; Zhou, H.L.; Lou, H.X.; Ren, D.M.; Shen, T. The genus Litsea in traditional Chinese medicine: An ethnomedical, phytochemical and pharmacological Review. J. Ethnopharmacol. 2015, 164, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Chanderbali, A.S.; Werff, H.; Renner, S.S. Phylogeny and historical biogeography of Lauraceae: Evidence from the chloroplast and nuclear genomes. Ann. Mo. Bot. Gard. 2001, 88, 104–134. [Google Scholar] [CrossRef]
- Tyagi, A.K.; Malik, A. Antimicrobial potential and chemical composition of eucalyptus globulus oil in liquid and vapour phase against food spoilage microorganisms. Food Chem. 2011, 126, 228–235. [Google Scholar] [CrossRef]
- Choudhury, S.; Ahmed, R.; Barthel, A.; Leclercq, P.A.; Leclercq, P.A. Composition of the stem, flower and fruit oils of Litsea cubeba pers. from two locations of Assam, India. J. Essent. Oil Res. 1998, 10, 381–386. [Google Scholar] [CrossRef]
- Kajaria, D.K.; Gangwar, M.; Kumar, D.; Sharma, A.K.; Tilak, R.; Nath, G.; Tripathi, Y.B.; Tripathi, J.S.; Tiwari, S.K. Evaluation of antimicrobial activity and bronchodialator effect of a polyherbal Drug-Shrishadi. Asian Pac. J. Trop. Biomed. 2012, 2, 905–909. [Google Scholar] [CrossRef]
- Kamle, M.; Mahato, D.K.; Lee, K.E.; Bajpai, V.K.; Gajurel, P.R.; Gu, K.S.; Kumar, P. Ethnopharmacological properties and medicinal uses of Litsea cubeba. Plants 2019, 8, 150. [Google Scholar] [CrossRef]
- Deng, Y.; Luo, Y.; He, Y.; Qin, X.; Li, C.; Deng, X. Complete chloroplast genome of Michelia shiluensis and a comparative analysis with four Magnoliaceae species. Forests 2020, 11, 267. [Google Scholar] [CrossRef]
- Yang, Z.; Zhao, T.; Ma, Q.; Liang, L.; Wang, G. Comparative genomics and phylogenetic analysis revealed the chloroplast genome variation and interspecific relationships of Corylus (Betulaceae) species. Front. Plant Sci. 2018, 9, 927. [Google Scholar] [CrossRef]
- Song, W.; Chen, Z.; He, L.; Feng, Q.; Zhang, H.; Du, G.; Shi, C.; Wang, S. Comparative chloroplast genome analysis of wax gourd (Benincasa hispida) with three Benincaseae species, revealing evolutionary dynamic patterns and phylogenetic implications. Genes 2022, 13, 461. [Google Scholar] [CrossRef]
- Krause, K. Piecing together the puzzle of parasitic plant plastome evolution. Planta 2011, 234, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.T.; Tran, N.; Nguyen, T.D.; Vu, Q.L.; Bui, M.H.; Le, M.T.; Le, L. Complete chloroplast genome of Paphiopedilum delenatii and phylogenetic relationships among Orchidaceae. Plants 2020, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Guo, L.; Zhao, W.; Xu, J.; Li, Y.; Zhang, X.; Shen, X.; Wu, M.; Hou, X. Complete chloroplast genome sequence and phylogenetic analysis of Paeonia ostii. Molecules 2018, 23, 246. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Xue, Q.; Wang, H.; Xie, X.; Zhu, S.; Liu, W.; Ding, X. Mutational biases and GC-biased gene conversion affect GC content in the plastomes of Dendrobium genus. Int. J. Mol. Sci. 2017, 18, 2307. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Han, L.; Chen, C.; Wang, Z. The Complete Chloroplast genome sequence of Epipremnum aureum and its comparative analysis among eight Araceae species. PLoS ONE 2018, 13, e0192956. [Google Scholar] [CrossRef]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; DePamphilis, C.W.; Chumley, T.W.; Haberle, R.C.; Wyman, S.K.; Alverson, A.J.; Peery, R.; Herman, S.J.; et al. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, S.; Liu, Y.; Yuan, Q.; Sun, J.; Guo, L. Chloroplast genome variation and phylogenetic relationships of Atractylodes species. BMC Genom. 2021, 22, 103. [Google Scholar] [CrossRef]
- Ju, J.; Xie, Y.; Yu, H.; Guo, Y.; Cheng, Y.; Zhang, R.; Yao, W. Major components in Lilac and Litsea cubeba essential oils kill Penicillium roqueforti through mitochondrial apoptosis pathway. Ind. Crops Prod. 2020, 149, 112349. [Google Scholar] [CrossRef]
- Liu, X.; Xu, X.; Zhao, J. A new generalized P-Value approach for testing equality of coefficients of variation in k normal Populations. J. Stat. Comput. Simul. 2011, 81, 1121–1130. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 September 2021).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de Novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De Novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. TRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Lehwark, P.; Greiner, S. GB2sequin—A file converter preparing custom GenBank files for database submission. Genomics 2019, 111, 759–761. [Google Scholar] [CrossRef]
- Zheng, S.; Poczai, P.; Hyvönen, J.; Tang, J.; Amiryousefi, A. Chloroplot: An online program for the versatile plotting of organelle genomes. Front. Genet. 2020, 11, 1123. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-Web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JmodelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Saski, C.; Lee, S.-B.; Daniell, H.; Wood, T.C.; Tomkins, J.; Kim, H.-G.; Jansen, R.K. Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol. Biol. 2005, 59, 309–322. [Google Scholar] [CrossRef]
- Cui, Y.; Nie, L.; Sun, W.; Xu, Z.; Wang, Y.; Yu, J.; Song, J.; Yao, H. Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family Zingiberaceae based on the complete chloroplast genome. Plants 2019, 8, 283. [Google Scholar] [CrossRef]
- McInerney, J. GCUA: General codon usage analysis. Bioinformatics 1998, 14, 372–373. [Google Scholar] [CrossRef]
- Zuo, L.H.; Shang, A.Q.; Zhang, S.; Yu, X.Y.; Ren, Y.C.; Yang, M.S.; Wang, J.M. The first complete chloroplast genome sequences of Ulmus species by de Novo sequencing: Genome comparative and taxonomic position analysis. PLoS ONE 2017, 12, e0171264. [Google Scholar] [CrossRef]
- Suo, Z.; Jin, X.; Suo, Z.L.; Li, W.Y.; Jin, X.B.; Zhang, H.J. A new nuclear DNA marker revealing both microsatellite variations and single nucleotide polymorphic loci: A case study on classification of cultivars in Lagerstroemia indica. Artic. J. Microb. Biochem. Technol. 2016, 8, 266–271. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, L.; Liu, A.; Chen, J.; Wu, L.; Hu, W.; Zhang, W.; Kim, K.; Lee, S.C.; Yang, T.J.; et al. The complete chloroplast genome sequences of five Epimedium species: Lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef]
- Chen, J.; Hao, Z.; Xu, H.; Yang, L.; Liu, G.; Sheng, Y.; Zheng, C.; Zheng, W.; Cheng, T.; Shi, J. The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front. Plant Sci. 2015, 6, 447. [Google Scholar] [CrossRef]
- Dong, W.L.; Wang, R.N.; Zhang, N.Y.; Fan, W.B.; Fang, M.F.; Li, Z.H. Molecular evolution of chloroplast genomes of orchid species: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef]
- Park, I.; Yang, S.; Choi, G.; Kim, W.J.; Cheol, M.B. The complete chloroplast genome sequences of Aconitum pseudolaeve and Aconitum longecassidatum, and development of molecular markers for distinguishing species in the Aconitum subgenus Lycoctonum. Molecules 2017, 22, 2012. [Google Scholar] [CrossRef] [Green Version]
- Mehmood, F.; Abdullah; Shahzadi, I.; Ahmed, I.; Waheed, M.T.; Mirza, B. Characterization of Withania somnifera chloroplast genome and its comparison with other selected species of Solanaceae. Genomics 2020, 112, 1522–1530. [Google Scholar] [CrossRef]
- Henriquez, C.L.; Abdullah; Ahmed, I.; Carlsen, M.M.; Zuluaga, A.; Croat, T.B.; McKain, M.R. Evolutionary dynamics of chloroplast genomes in subfamily Aroideae (Araceae). Genomics 2020, 112, 2349–2360. [Google Scholar] [CrossRef]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef]
- Huo, Y.; Gao, L.; Liu, B.; Yang, Y.; Kong, S.; Sun, Y.; Yang, Y.; Wu, X. Complete chloroplast genome sequences of four Allium species: Comparative and phylogenetic analyses. Sci. Rep. 2019, 9, 12250. [Google Scholar] [CrossRef]
- Muraguri, S.; Xu, W.; Chapman, M.; Muchugi, A.; Oluwaniyi, A.; Oyebanji, O.; Liu, A. Intraspecific variation within castor bean (Ricinus communis L.) based on chloroplast genomes. Ind. Crops Prod. 2020, 155, 112779. [Google Scholar] [CrossRef]
- Kim, Y.; Shin, J.; Oh, D.R.; Kim, A.Y.; Choi, C. Comparative analysis of complete chloroplast genome sequences and insertion-deletion (Indel) polymorphisms to distinguish five Vaccinium species. Forests 2020, 11, 927. [Google Scholar] [CrossRef]
- Song, W.; Ji, C.; Chen, Z.; Cai, H.; Wu, X.; Shi, C.; Wang, S. Comparative analysis the complete chloroplast genomes of nine Musa species: Genomic features, comparative analysis, and phylogenetic implications. Front. Plant Sci. 2022, 13, 62. [Google Scholar] [CrossRef]
- Tian, S.; Lu, P.; Zhang, Z.; Wu, J.Q.; Zhang, H.; Shen, H. Chloroplast genome sequence of chongming lima bean (Phaseolus lunatus L.) and comparative analyses with other legume chloroplast genomes. BMC Genomics 2021, 22, 194. [Google Scholar] [CrossRef]
- Alzahrani, D.A.; Yaradua, S.S.; Yaradua, S.S.; Albokhari, E.J.; Albokhari, E.J.; Abba, A. Complete chloroplast genome sequence of Barleria prionitis, comparative chloroplast genomics and phylogenetic relationships among Acanthoideae. BMC Genomics 2020, 21, 393. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, T.; Bai, G.; Zhao, Y. Complete chloroplast genome sequence of Fagopyrum dibotrys: Genome features, comparative analysis and phylogenetic relationships. Sci. Rep. 2018, 8, 12379. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Du, J.; Gao, L.; Li, Y.; Hou, X. The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): Genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene 2019, 699, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zheng, Y. Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Sci. Rep. 2018, 8, 9285. [Google Scholar] [CrossRef] [PubMed]
- Zong, D.; Gan, P.; Zhou, A.; Zhang, Y.; Zou, X.; Duan, A.; Song, Y.; He, C. Plastome sequences help to resolve deep-level relationships of Populus in the family Salicaceae. Front. Plant Sci. 2019, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.-Y.; Gao, L.Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef]
- Meng, K.K.; Chen, S.F.; Xu, K.W.; Zhou, R.C.; Li, M.W.; Dhamala, M.K.; Liao, W.B.; Fan, Q. Phylogenomic analyses based on genome-skimming data reveal cyto-nuclear Discordance in the eolutionary history of Cotoneaster (Rosaceae). Mol. Phylogenet. Evol. 2021, 158, 107083. [Google Scholar] [CrossRef]
- Li, P.; Lu, R.S.; Xu, W.Q.; Ohi-Toma, T.; Cai, M.Q.; Qiu, Y.X.; Cameron, K.M.; Fu, C.X. Comparative genomics and phylogenomics of East Asian tulips (Amana, Liliaceae). Front. Plant Sci. 2017, 10, 451. [Google Scholar] [CrossRef]
- Nater, A.; Burri, R.; Kawakami, T.; Smeds, L.; Ellegren, H. Resolving evolutionary relationships in closely related species with whole-genome sequencing data. Syst. Biol. 2015, 64, 1000. [Google Scholar] [CrossRef]
- Ma, J.; Clemants, S. A history and overview of the Flora Reipublicae Popularis Sinicae (FRPS, Flora of China, Chinese Edition, 1959–2004). Taxon 2006, 55, 451–460. [Google Scholar] [CrossRef]
- Sang, T. Utility of low-copy nuclear gene sequences in plant phylogenetics. Crit. Rev. Biochem. Mol. 2002, 37, 121–147. [Google Scholar] [CrossRef]
- Li, J.; Christophel, D.C.; Conran, J.G.; Li, H.W. Phylogenetic Relationships within the “core” Laureae (Litsea Complex, Lauraceae) Inferred from Sequences of the Chloroplast Gene MatK and Nuclear Ribosomal DNA ITS Regions. Plant Syst. Evol. 2004, 246, 19–34. [Google Scholar] [CrossRef]
- Fijridiyanto, I.A.; Murakami, N. Phylogeny of Litsea and Related Genera (Laureae-Lauraceae) based on analysis of Rpb2 gene sequences. J. Plant Res. 2009, 122, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, S.G.; Krishnamurthy, Y.L.; Kumar, S.S. Diversity, ecology and molecular phylogeny of genus Litsea (Lauraceae) in central western ghat areas of India. Trop. Ecol. 2021, 62, 644–652. [Google Scholar] [CrossRef]
- Li, H.T.; Yi, T.S.; Gao, L.M.; Ma, P.F.; Zhang, T.; Yang, J.B.; Gitzendanner, M.A.; Fritsch, P.W.; Cai, J.; Luo, Y.; et al. Origin of angiosperms and the puzzle of the Jurassic Gap. Nat. Plants 2019, 5, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tian, Y.; Tng, D.Y.P.; Zhou, J.; Zhang, Y.; Wang, Z.; Li, P.; Wang, Z. Comparative chloroplast genomics of Litsea Lam. (Lauraceae) and its phylogenetic implications. Forests 2021, 12, 744. [Google Scholar] [CrossRef]
- Abou-Shaara, H.F.; Syed Abbas, A.; AL-Kahtani, S.N.; Taha, E.K.A.; Ali Khan, K.; Jamal, Z.A.; Alhumaidi Alotaibi, M.; Ahmad, B.; Ahmad Khan, N.; Qamer, S.; et al. Exploring the Non-Coding regions in the MtDNA of some honey bee species and subspecies. Saudi J. Biol. Sci. 2021, 28, 204–209. [Google Scholar] [CrossRef]
- Wu, L.W.; Cui, Y.X.; Wang, Q.; Xu, Z.C.; Wang, Y.; Lin, Y.L.; Song, J.Y.; Yao, H. Identification and phylogenetic analysis of five Crataegus species (Rosaceae) based on complete chloroplast genomes. Planta 2021, 254, 14. [Google Scholar] [CrossRef]
- Liu, Z.F.; Ma, H.; Ci, X.Q.; Li, L.; Song, Y.; Liu, B.; Li, H.W.; Wang, S.L.; Qu, X.J.; Hu, J.L.; et al. Can plastid genome sequencing be used for species identification in Lauraceae? Bot. J. Linn. Soc. 2021, 197, e7662. [Google Scholar] [CrossRef]
- Chen, Y.C.; Li, Z.; Zhao, Y.X.; Gao, M.; Wang, J.Y.; Liu, K.W.; Wang, X.; Wu, L.W.; Jiao, Y.L.; Xu, Z.L.; et al. The Litsea genome and the evolution of the laurel family. Nat. Commun. 2020, 11, 1675. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Genes | Gene Names | Amount |
|---|---|---|
| Pholosystem I | psaA, psaB, psaC, psal, psaJ | 5 |
| Photosystem II | psbA, psbK, psbl, psbM, psbD, psbC, psbZ, psbG, psbL, psbF, psbE, psbB, psbT, psbN, psbH | 15 |
| Cytochrome | petA, petG, petL, petN, petB, petD | 6 |
| ATP syntliase | atpA, atpF, atpH, atpI, atpE, atpB | 6 |
| NADH dehydrogenase | ndhJ, ndhB *, ndhK, ndhC, ndhD, ndhF, ndhE, ndhG, ndhl, ndhA, ndhH | 12 |
| RubisCO large subunit | rbcL | 1 |
| RNA polymerase | RpoCl, rpoC2, rpoB, rpoA | 4 |
| Ribosomal proteins (SSU) | rps16, rpsl2 *, rps2, rps14, rps4, rps18, rps7 *, rps11, rps8, rps3, rps19, rps15 | 14 |
| Ribosomal proteins (LSU) | rpl33, rpl20, rpl36, rpll4, rpll6, rpl22, rpl2, rpl23, rpl32 | 9 |
| Transfer RNAs | trnH-GUG, trnK-UUU, trnQ-UUG, trnS-GCU, trnG-UCC, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnT-GGU, trnS-UGA, trnG-UCC, trnT-GGU, trnS-UGA, trnG-UCQ, trnM-CAU, trnS-GGA, trnT-UGU, trnL-UAA, trnF-GUU, trnV-UAC, trnM-CAU, trnW-CCA, trnP-UGG, trnl-CAU, trnA-UGC, trnR-ACG, trnL-UAG, trnN-GUU, trnR-GUG, trnA-UGC, trnl-GAU, trnL-CAA | 34 |
| Ribosomal RNAs | rrn4.5 *, rrn5 *, rrn16 *, rrn23 * | 8 |
| Hypothetical chloroplast reading frames (ycf) | ycfl, ycf2, ycf3, ycf4 | 4 |
| Other genes | accD, clpP, ccsA, cemA, infA, rpoA, matK | 7 |
| L. auriculata | L. chunii | L. ichangensis | L. moupinensis | L. populifolia | L. rubescens | L. sericea | L. tsinlingensis | L. veitchiana |
|---|---|---|---|---|---|---|---|---|
| 152,377 | 152,081 | 152,747 | 152,588 | 152,619 | 152,581 | 152,717 | 152,051 | 152,578 |
| 93,535 | 93,138 | 93,631 | 93,552 | 93,569 | 93,550 | 93,583 | 93,093 | 93,540 |
| 18,814 | 18,813 | 18,902 | 18,824 | 18,838 | 18,819 | 18,900 | 18,828 | 18,826 |
| 20,014 | 20,065 | 20,107 | 20,106 | 20,106 | 20,106 | 20,117 | 20,065 | 20,106 |
| 39.2 | 39.2 | 39.2 | 39.2 | 39.2 | 39.2 | 39.1 | 39.2 | 39.2 |
| 37.9 | 37.9 | 38.0 | 37.9 | 38.0 | 37.9 | 37.9 | 37.9 | 38.0 |
| 33.9 | 33.9 | 33.9 | 33.9 | 33.9 | 33.9 | 33.9 | 34.0 | 34.0 |
| 44.4 | 44.4 | 44.4 | 44.4 | 44.4 | 44.4 | 44.4 | 44.4 | 44.4 |
| 128 (113) | 128 (113) | 128 (113) | 128 (113) | 128 (113) | 128 (113) | 128 (113) | 128 (113) | 128 (113) |
| 84 (79) | 84 (79) | 84 (79) | 84 (79) | 84 (79) | 84 (79) | 84 (79) | 84 (79) | 84 (79) |
| 9 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) | 8 (4) |
| 36 (30) | 36 (30) | 36 (30) | 36 (30) | 36 (30) | 36 (30) | 36 (30) | 36 (30) | 36 (30) |
| NC_056809 | NC_056810 | NC_056811 | NC_056812 | NC_056813 | NC_056814 | NC_056815 | NC_056816 | NC_056817 |
| 49.12 | 49.35 | 49.06 | 49.12 | 49.11 | 49.12 | 49.15 | 49.37 | 49.12 |
| 8.51 | 7.54 | 8.81 | 8.82 | 8.82 | 8.95 | 8.81 | 8.85 | 8.82 |
| 30.19 | 29.92 | 30.93 | 30.85 | 30.86 | 1.36 | 30.83 | 29.90 | 30.85 |
| 13 | 13 | 13 | 13 | 13 | 13 | 13 | 13 | 13 |
| Species | Region | Transition Substitutions | Transversion Substitutions | ||||
|---|---|---|---|---|---|---|---|
| A/G | C/T | A/T | A/C | C/G | G/T | ||
| L. auriculata | 109 | 106 | 25 | 46 | 6 | 63 | |
| L. chunii | Large | 137 | 129 | 20 | 52 | 11 | 80 |
| L. ichangensis | 129 | 139 | 26 | 58 | 11 | 75 | |
| L. moupinensis | single | 134 | 139 | 21 | 58 | 10 | 78 |
| L. populifolia | copy | 129 | 138 | 22 | 60 | 10 | 80 |
| L. rubescens | 134 | 140 | 22 | 58 | 10 | 78 | |
| L. sericea | 123 | 129 | 23 | 55 | 10 | 78 | |
| L. tsinlingensis | 136 | 127 | 19 | 56 | 11 | 73 | |
| L. veitchiana | 127 | 128 | 21 | 59 | 10 | 75 | |
| L. auriculata | 3 | 5 | 0 | 2 | 2 | 11 | |
| L. chunii | Inverted repeat | 4 | 8 | 2 | 12 | 1 | 15 |
| L. ichangensis | 4 | 5 | 1 | 3 | 1 | 3 | |
| L. moupinensis | 3 | 8 | 2 | 12 | 1 | 12 | |
| L. populifolia | 3 | 6 | 2 | 12 | 1 | 11 | |
| L. rubescens | 3 | 8 | 2 | 12 | 1 | 11 | |
| L. sericea | 3 | 6 | 3 | 12 | 1 | 12 | |
| L. tsinlingensis | 2 | 9 | 2 | 12 | 1 | 14 | |
| L. veitchiana | 3 | 7 | 2 | 12 | 1 | 12 | |
| L. auriculata | 43 | 47 | 5 | 21 | 3 | 16 | |
| L. chunii | Small | 42 | 37 | 10 | 19 | 4 | 17 |
| L. ichangensis | 42 | 45 | 5 | 24 | 5 | 21 | |
| L. moupinensis | 38 | 41 | 5 | 24 | 6 | 21 | |
| L. populifolia | single | 37 | 43 | 4 | 19 | 5 | 21 |
| L. rubescens | copy | 38 | 41 | 5 | 10 | 6 | 21 |
| L. sericea | 38 | 39 | 6 | 19 | 5 | 24 | |
| L. tsinlingensis | 42 | 37 | 5 | 20 | 4 | 18 | |
| L. veitchiana | 37 | 43 | 4 | 18 | 5 | 22 | |
| Comparative Analyses of InDel Sites | ||||||
|---|---|---|---|---|---|---|
| Species | Large Single Copy | Inverted Repeat | Small Single Copy | |||
| No′s of InDels | InDels′ Average Length (bp) | No′s of InDels | InDels′ Average Length (bp) | No′s of InDels | InDels′ Average Length (bp) | |
| L. auriculata | 86 | 4.40 | 16 | 103.3 | 18 | 1.3 |
| L. chunii | 89 | 8.4 | 3 | 458.7 | 20 | 1.4 |
| L. ichangensis | 99 | 3.7 | 5 | 276.0 | 19 | 1.6 |
| L. moupinensis | 88 | 3.7 | 4 | 339.0 | 16 | 1.9 |
| L. populifolia | 86 | 3.8 | 4 | 339.0 | 17 | 1.5 |
| L. rubescens | 88 | 3.9 | 5 | 272.4 | 16 | 1.9 |
| L. sericea | 86 | 3.7 | 4 | 339.0 | 19 | 2.0 |
| L. tsinlingensis | 87 | 9.1 | 2 | 678.0 | 19 | 1.4 |
| L. veitchiana | 88 | 3.7 | 4 | 339.0 | 18 | 1.8 |
| Average | 88.6 | 4.90 | 5.2 | 349.4 | 18.0 | 1.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, W.; Chen, Z.; Shi, W.; Han, W.; Feng, Q.; Shi, C.; Engel, M.S.; Wang, S. Comparative Analysis of Complete Chloroplast Genomes of Nine Species of Litsea (Lauraceae): Hypervariable Regions, Positive Selection, and Phylogenetic Relationships. Genes 2022, 13, 1550. https://doi.org/10.3390/genes13091550
Song W, Chen Z, Shi W, Han W, Feng Q, Shi C, Engel MS, Wang S. Comparative Analysis of Complete Chloroplast Genomes of Nine Species of Litsea (Lauraceae): Hypervariable Regions, Positive Selection, and Phylogenetic Relationships. Genes. 2022; 13(9):1550. https://doi.org/10.3390/genes13091550
Chicago/Turabian StyleSong, Weicai, Zimeng Chen, Wenbo Shi, Weiqi Han, Qi Feng, Chao Shi, Michael S. Engel, and Shuo Wang. 2022. "Comparative Analysis of Complete Chloroplast Genomes of Nine Species of Litsea (Lauraceae): Hypervariable Regions, Positive Selection, and Phylogenetic Relationships" Genes 13, no. 9: 1550. https://doi.org/10.3390/genes13091550