
Identification of Ancestry Informative Markers in Mediterranean Trout Populations of Molise (Italy): A Multi-Methodological Approach with Machine Learning

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Filtering Procedures and Reference Dataset Building
2.2. Marker Selection
2.3. Validation Dataset
2.4. SNP Annotation
3. Results
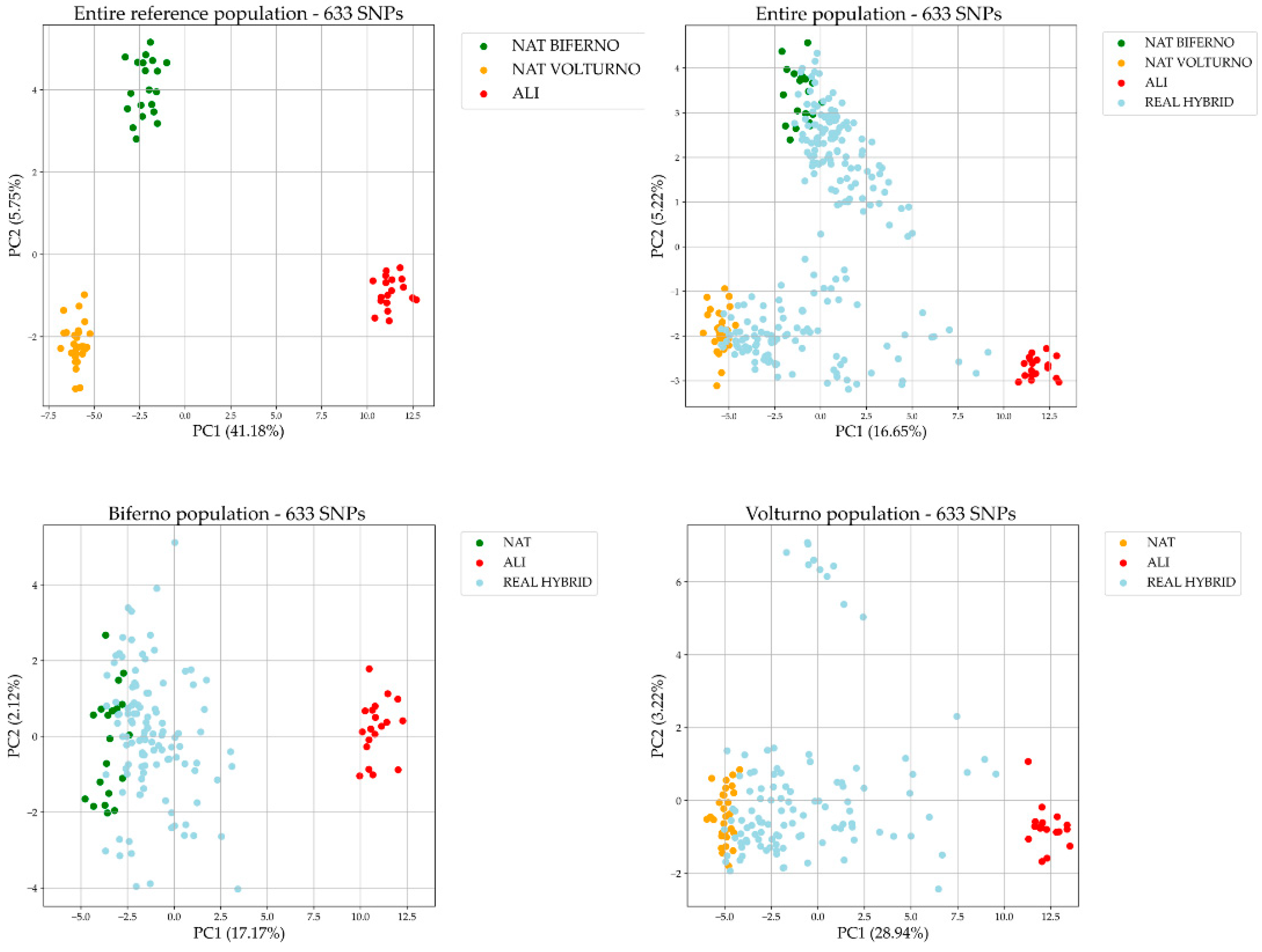
3.1. Population Overview
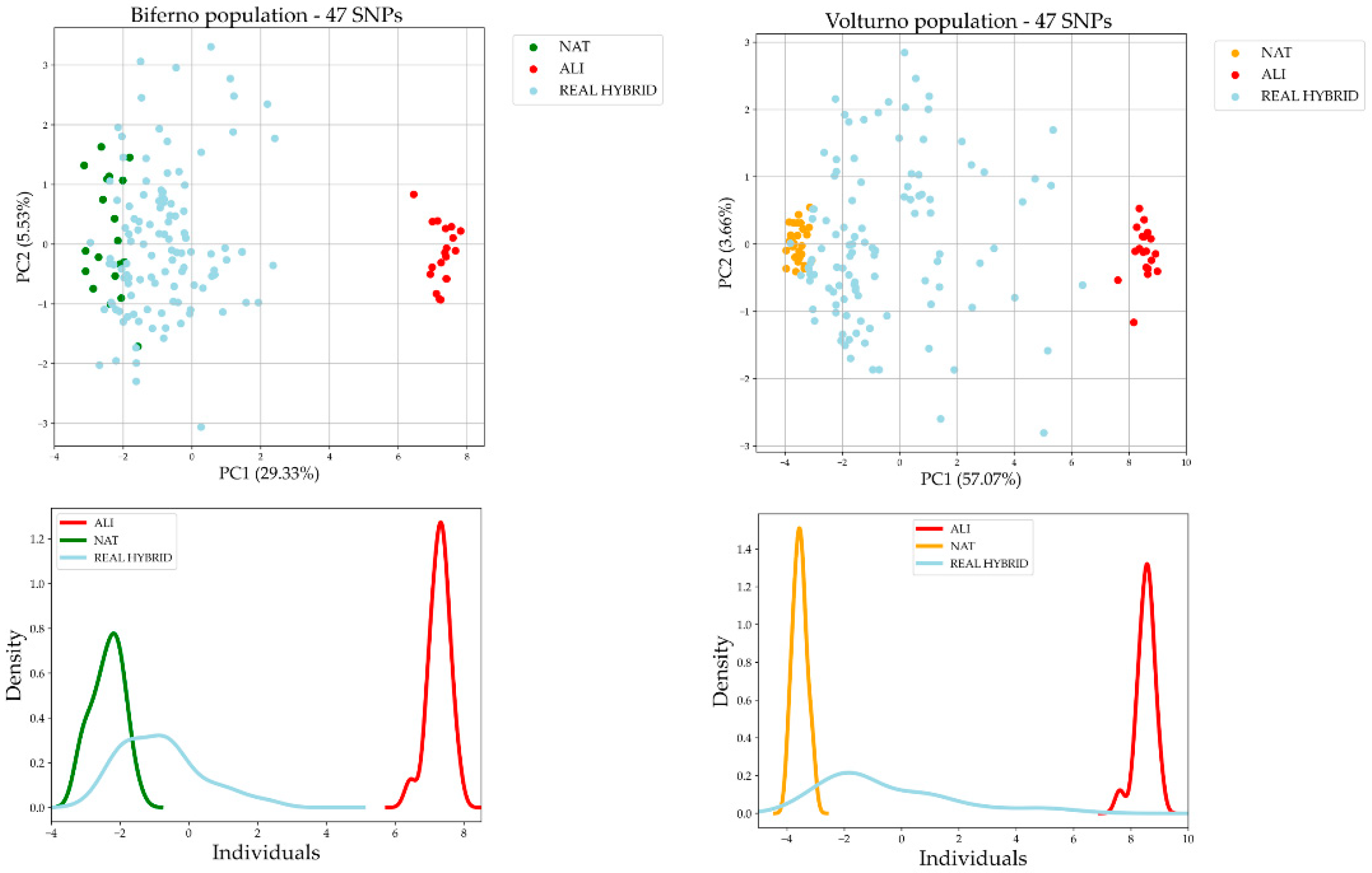
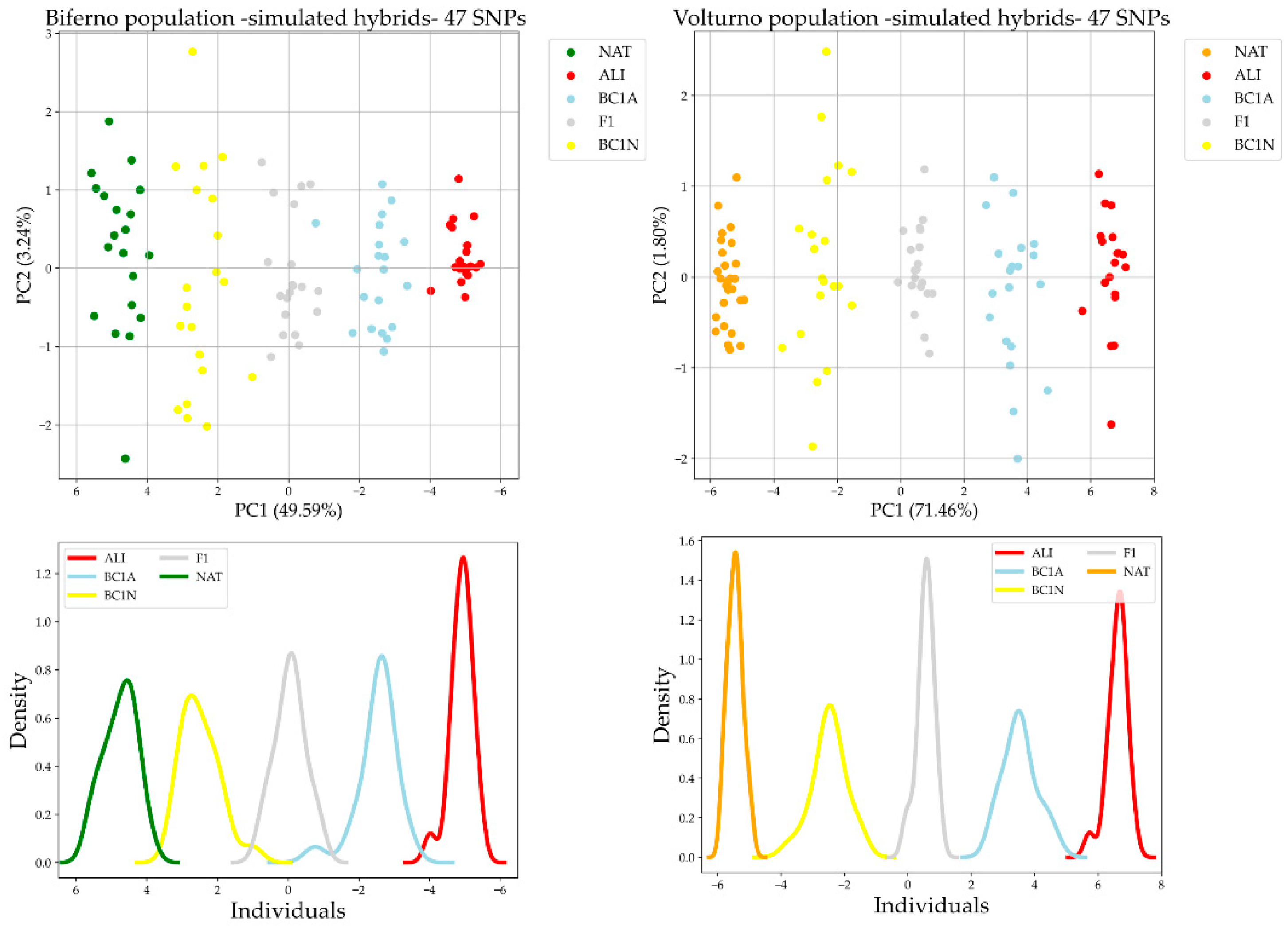
3.2. Comparison of AIMs Selection Methods and Validation
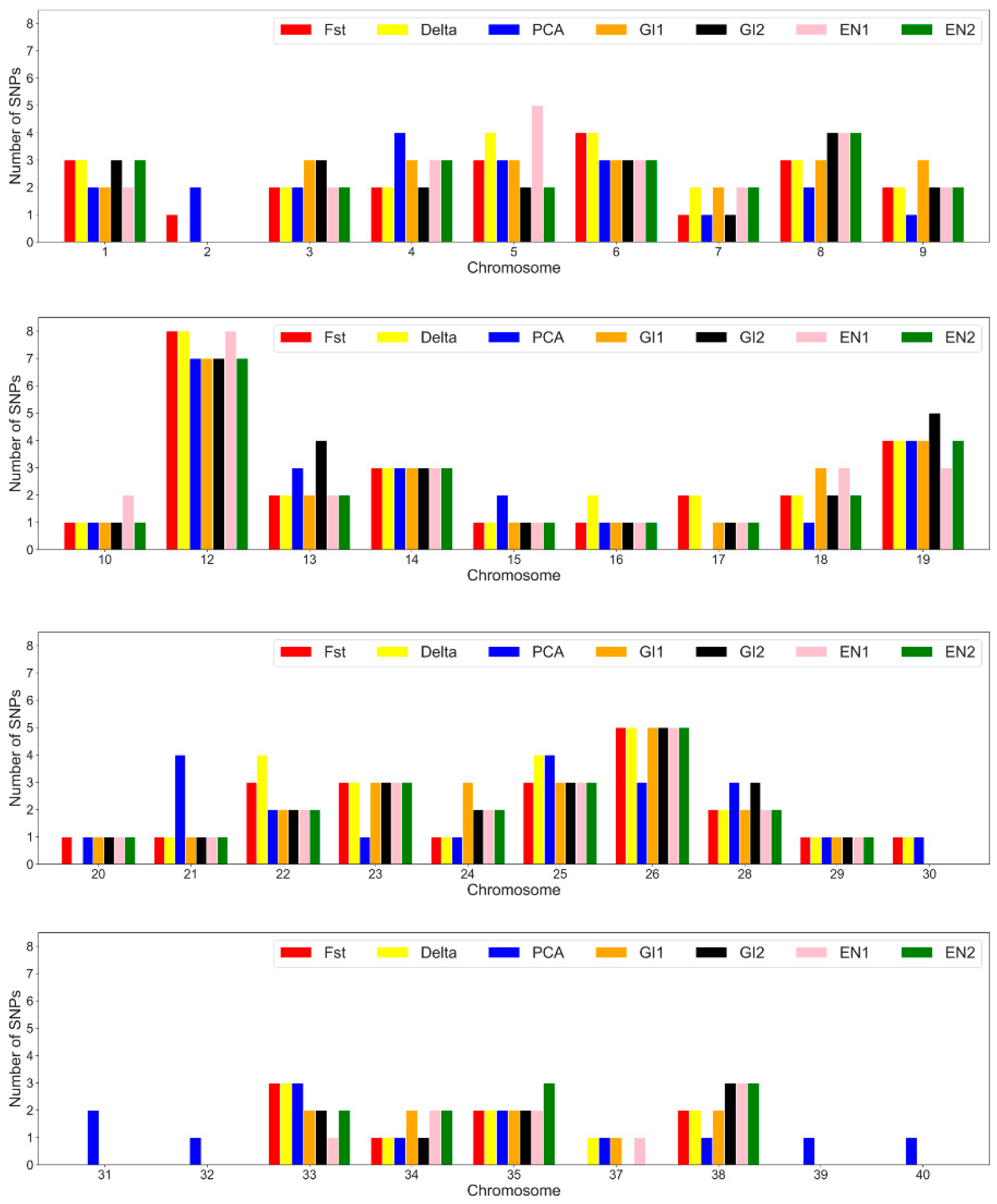
3.3. SNP Annotation and Marked Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strayer, D.L. Alien species in fresh waters: Ecological effects, interactions with other stressors, and prospects for the future. Freshw. Biol. 2010, 55, 152–174. [Google Scholar] [CrossRef]
- Gurevitch, J.; Padilla, D.K. Are invasive species a major cause of extinctions? Trends Ecol. Evol. 2004, 19, 470–474. [Google Scholar] [CrossRef]
- Bernatchez, L. The Evolutionary History of Brown Trout (Salmo trutta L.) Inferred from Phylogeographic, Nested Clade, and Mismatch Analyses of Mitochondrial DNA Variation. Evolution 2001, 55, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Hashemzadeh Segherloo, I.; Freyhof, J.; Berrebi, P.; Ferchaud, A.-L.; Geiger, M.; Laroche, J.; Levin, B.A.; Normandeau, E.; Bernatchez, L. A genomic perspective on an old question: Salmo trouts or Salmo trutta (Teleostei: Salmonidae)? Mol. Phylogenet. Evol. 2021, 162, 107204. [Google Scholar] [CrossRef]
- Rossi, A.R.; Talarico, L.; Petrosino, G.; Crescenzo, S.; Tancioni, L. Conservation Genetics of Mediterranean Brown Trout in Central Italy (Latium): A Multi-Marker Approach. Water 2022, 14, 937. [Google Scholar] [CrossRef]
- Palombo, V.; De Zio, E.; Salvatore, G.; Esposito, S.; Iaffaldano, N.; D’Andrea, M. Genotyping of Two Mediterranean Trout Populations in Central-Southern Italy for Conservation Purposes Using a Rainbow-Trout-Derived SNP Array. Animals 2021, 11, 1803. [Google Scholar] [CrossRef]
- Sabatini, A.; Podda, C.; Frau, G.; Cani, M.V.; Musu, A.; Serra, M.; Palmas, F. Restoration of native Mediterranean brown trout Salmo cettii Rafinesque, 1810 (Actinopterygii: Salmonidae) populations using an electric barrier as a mitigation tool. Eur. Zool. J. 2018, 85, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.R.; Petrosino, G.; Milana, V.; Martinoli, M.; Rakaj, A.; Tancioni, L. Genetic identification of native populations of Mediterranean brown trout Salmo trutta L. complex (Osteichthyes: Salmonidae) in central Italy. Eur. Zool. J. 2019, 86, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Lorenzoni, M.; Carosi, A.; Giovannotti, M.; Porta, G.L.; Splendiani, A.; Barucchi, V.C. Ecology and conservation of the Mediterranean trout in the central Apennines (Italy). J. Limnol. 2019, 78, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Splendiani, A.; Giovannotti, M.; Righi, T.; Fioravanti, T.; Cerioni, P.N.; Lorenzoni, M.; Carosi, A.; La Porta, G.; Barucchi, V.C. Introgression despite protection: The case of native brown trout in Natura 2000 network in Italy. Conserv. Genet. 2019, 20, 343–356. [Google Scholar] [CrossRef]
- Lorenzoni, M.; Carosi, A.; Giovannotti, M.; Porta, G.L.; Splendiani, A.; Barucchi, V.C. Morphological survey as powerful detection tool of pure and local phenotypes in Salmo trutta complex. Knowl. Manag. Aquat. Ecosyst. 2019, 420, 48. [Google Scholar] [CrossRef]
- Abdul-Muneer, P.M. Application of microsatellite markers in conservation genetics and fisheries management: Recent advances in population structure analysis and conservation strategies. Genet. Res. Int. 2014, 2014, 691759. [Google Scholar] [CrossRef] [Green Version]
- Splendiani, A.; Giovannotti, M.; Cerioni, P.N.; Caniglia, M.L.; Caputo, V. Phylogeographic inferences on the native brown trout mtDNA variation in central Italy. Ital. J. Zool. 2006, 73, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Poteaux, C.; Bonhomme, F.; Berrebi, P. Microsatellite polymorphism and genetic impact of restocking in Mediterranean brown trout (Salmo trutta L.). Heredity 1999, 82, 645–653. [Google Scholar] [CrossRef]
- Saint-Pé, K.; Leitwein, M.; Tissot, L.; Poulet, N.; Guinand, B.; Berrebi, P.; Marselli, G.; Lascaux, J.-M.; Gagnaire, P.-A.; Blanchet, S. Development of a large SNPs resource and a low-density SNP array for brown trout (Salmo trutta) population genetics. BMC Genom. 2019, 20, 582. [Google Scholar] [CrossRef] [Green Version]
- Segelbacher, G.; Bosse, M.; Burger, P.; Galbusera, P.; Godoy, J.A.; Helsen, P.; Hvilsom, C.; Iacolina, L.; Kahric, A.; Manfrin, C.; et al. New developments in the field of genomic technologies and their relevance to conservation management. Conserv. Genet. 2022, 23, 217–242. [Google Scholar] [CrossRef]
- Casanova Chiclana, A. Population Genomics as a Tool for Management and Conservation of Brown Trout (Salmo trutta) in the Iberian Peninsula. Ph.D. Thesis, Universidade de Santiago de Compostela, Santiago de Compostela, Spain, 2021. [Google Scholar]
- Havlick, D.G.; Biermann, C. Wild, Native, or Pure: Trout as Genetic Bodies. Sci. Technol. Hum. Values 2021, 46, 1201–1229. [Google Scholar] [CrossRef]
- Drywa, A.; Poćwierz-Kotus, A.; Dobosz, S.; Kent, M.P.; Lien, S.; Wenne, R. Identification of multiple diagnostic SNP loci for differentiation of three salmonid species using SNP-arrays. Mar. Genom. 2014, 15, 5–6. [Google Scholar] [CrossRef]
- D’Agaro, E.; Gibertoni, P.; Marroni, F.; Messina, M.; Tibaldi, E.; Esposito, S. Genetic and Phenotypic Characteristics of the Salmo trutta Complex in Italy. Appl. Sci. 2022, 12, 3219. [Google Scholar] [CrossRef]
- Lobón-Cerviá, J.; Sanz, N. Brown Trout: Biology, Ecology and Management; John Wiley & Sons Inc.: Chichester, UK, 2017; ISBN 978-1-119-26831-4. [Google Scholar]
- Wenne, R.; Drywa, A.; Kent, M.; Sundsaasen, K.K.; Lien, S. SNP Arrays for Species Identification in Salmonids. In Marine Genomics: Methods and Protocols; Bourlat, S.J., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 97–111. ISBN 978-1-4939-3774-5. [Google Scholar]
- Palti, Y.; Gao, G.; Liu, S.; Kent, M.P.; Lien, S.; Miller, M.R.; Rexroad, C.E.; Moen, T. The development and characterization of a 57K single nucleotide polymorphism array for rainbow trout. Mol. Ecol. Resour. 2015, 15, 662–672. [Google Scholar] [CrossRef]
- Lao, O.; van Duijn, K.; Kersbergen, P.; de Knijff, P.; Kayser, M. Proportioning whole-genome single-nucleotide-polymorphism diversity for the identification of geographic population structure and genetic ancestry. Am. J. Hum. Genet. 2006, 78, 680–690. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, F.; Galimberti, G.; Schiavo, G.; Mastrangelo, S.; Di Gerlando, R.; Strillacci, M.G.; Bagnato, A.; Portolano, B.; Fontanesi, L. Preselection statistics and Random Forest classification identify population informative single nucleotide polymorphisms in cosmopolitan and autochthonous cattle breeds. Animal 2018, 12, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Bertolini, F.; Galimberti, G.; Bovo, S.; Dall’Olio, S.; Nanni Costa, L.; Gallo, M.; Fontanesi, L. A machine learning approach for the identification of population-informative markers from high-throughput genotyping data: Application to several pig breeds. Animal 2020, 14, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, I.; Henriques, D.; Johnston, J.S.; Chávez-Galarza, J.; Kryger, P.; Pinto, M.A. Reduced SNP Panels for Genetic Identification and Introgression Analysis in the Dark Honey Bee (Apis mellifera mellifera). PLoS ONE 2015, 10, e0124365. [Google Scholar] [CrossRef] [Green Version]
- Stronen, A.V.; Mattucci, F.; Fabbri, E.; Galaverni, M.; Cocchiararo, B.; Nowak, C.; Godinho, R.; Ruiz-González, A.; Kusak, J.; Skrbinšek, T.; et al. A reduced SNP panel to trace gene flow across southern European wolf populations and detect hybridization with other Canis taxa. Sci. Rep. 2022, 12, 4195. [Google Scholar] [CrossRef]
- Kavakiotis, I.; Triantafyllidis, A.; Ntelidou, D.; Alexandri, P.; Megens, H.-J.; Crooijmans, R.P.M.A.; Groenen, M.A.M.; Tsoumakas, G.; Vlahavas, I. TRES: Identification of Discriminatory and Informative SNPs from Population Genomic Data. J. Hered. 2015, 106, 672–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somenzi, E.; Ajmone-Marsan, P.; Barbato, M. Identification of Ancestry Informative Marker (AIM) Panels to Assess Hybridisation between Feral and Domestic Sheep. Animals 2020, 10, 582. [Google Scholar] [CrossRef] [Green Version]
- McMeel, O.M.; Hoey, E.M.; Ferguson, A. Partial nucleotide sequences, and routine typing by polymerase chain reaction–restriction fragment length polymorphism, of the brown trout (Salmo trutta) lactate dehydrogenase, LDH-C1*90 and *100 alleles. Mol. Ecol. 2001, 10, 29–34. [Google Scholar] [CrossRef]
- Chiesa, S.; Filonzi, L.; Ferrari, C.; Vaghi, M.; Bilò, F.; Piccinini, A.; Zuccon, G.; Wilson, R.C.; Ulheim, J.; Nonnis Marzano, F. Combinations of distinct molecular markers allow to genetically characterize marble trout (Salmo marmoratus) breeders and stocks suitable for reintroduction plans. Fish. Res. 2016, 176, 55–64. [Google Scholar] [CrossRef]
- Pensierini, M.; Nonnis Marzano, F.; Gandolfi, G.; Maldini, M.; Marconato, E.; Gibertoni, P. Genotypes and phenotypes of mediterranean brown trout: Molecular investigation combined to morphological characterization for identification of autochthonous specimens. Quaterni ETP-J. Freshw. Biol. 2006, 34, 69–75. [Google Scholar]
- Splendiani, A.; Ruggeri, P.; Giovannotti, M.; Pesaresi, S.; Occhipinti, G.; Fioravanti, T.; Lorenzoni, M.; Nisi Cerioni, P.; Caputo Barucchi, V. Alien brown trout invasion of the Italian peninsula: The role of geological, climate and anthropogenic factors. Biol. Invasions 2016, 18, 2029–2044. [Google Scholar] [CrossRef]
- Berrebi, P. Three brown trout Salmo trutta lineages in Corsica described through allozyme variation. J. Fish Biol. 2015, 86, 60–73. [Google Scholar] [CrossRef]
- Fabiani, A.; Gratton, P.; Zappes, I.A.; Seminara, M.; D’Orsi, A.; Sbordoni, V.; Allegrucci, G. Investigating the genetic structure of trout from the Garden of Ninfa (central Italy): Suggestions for conservation and management. Fish. Manag. Ecol. 2018, 25, 1–11. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Wilkinson, S.; Wiener, P.; Archibald, A.L.; Law, A.; Schnabel, R.D.; McKay, S.D.; Taylor, J.F.; Ogden, R. Evaluation of approaches for identifying population informative markers from high density SNP Chips. BMC Genet. 2011, 12, 45. [Google Scholar] [CrossRef] [Green Version]
- Paschou, P.; Ziv, E.; Burchard, E.G.; Choudhry, S.; Rodriguez-Cintron, W.; Mahoney, M.W.; Drineas, P. PCA-Correlated SNPs for Structure Identification in Worldwide Human Populations. PLOS Genet. 2007, 3, e160. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Palombo, V.; Milanesi, M.; Sferra, G.; Capomaccio, S.; Sgorlon, S.; D’Andrea, M. PANEV: An R package for a pathway-based network visualization. BMC Bioinform. 2020, 21, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimand, J.; Kull, M.; Peterson, H.; Hansen, J.; Vilo, J. g:Profiler—A web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007, 35, W193–W200. [Google Scholar] [CrossRef] [PubMed]
- Frkonja, A.; Gredler, B.; Schnyder, U.; Curik, I.; Sölkner, J. Prediction of breed composition in an admixed cattle population. Anim. Genet. 2012, 43, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Abas, Z.; Dadousis, C.; Lykidis, D.; Paschou, P.; Drineas, P. Tracing cattle breeds with principal components analysis ancestry informative SNPs. PLoS ONE 2011, 6, e18007. [Google Scholar] [CrossRef] [Green Version]
- Casanova, A.; Heras, S.; Abras, A.; Roldán, M.I.; Bouza, C.; Vera, M.; García-Marín, J.L.; Martínez, P. Genomic Hatchery Introgression in Brown Trout (Salmo trutta L.): Development of a Diagnostic SNP Panel for Monitoring the Impacted Mediterranean Rivers. Genes 2022, 13, 255. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.; Cho, S.; Manjula, P.; Choi, N.; Kim, Y.-K.; Koh, Y.J.; Lee, S.H.; Kim, H.-Y.; Lee, J.H. Identification of Target Chicken Populations by Machine Learning Models Using the Minimum Number of SNPs. Animals 2021, 11, 241. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, F.; Galimberti, G.; Calò, D.G.; Schiavo, G.; Matassino, D.; Fontanesi, L. Combined use of principal component analysis and random forests identify population-informative single nucleotide polymorphisms: Application in cattle breeds. J. Anim. Breed. Genet. 2015, 132, 346–356. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | RF GI 1 | RF GI 2 | RF EN 1 | RF EN 2 | Delta | FST | PCA |
|---|---|---|---|---|---|---|---|
| RF GI 1 | 96 | ||||||
| RF GI 2 | 83 | 96 | |||||
| RF EN 1 | 89 | 81 | 96 | ||||
| RF EN 2 | 85 | 89 | 84 | 96 | |||
| Delta | 79 | 77 | 80 | 80 | 96 | ||
| FST | 81 | 81 | 80 | 83 | 88 | 96 | |
| PCA | 52 | 50 | 53 | 53 | 56 | 57 | 96 |
| Method | OOB Score | Train Accuracy | Test Accuracy |
|---|---|---|---|
| RF GI 1 | 90% | 100% | 95% |
| RF GI 2 | 87% | 100% | 97% |
| RF EN 1 | 85% | 100% | 95% |
| RF EN 2 | 86% | 100% | 97% |
| Delta | 91% | 100% | 92% |
| FST | 87% | 100% | 92% |
| PCA | 87% | 100% | 95% |
| SNPs Panel | N | Biferno (r2) | Volturno (r2) |
|---|---|---|---|
| Delta | 96 | 0.982 | 0.989 |
| FST | 96 | 0.981 | 0.988 |
| PCA | 96 | 0.973 | 0.984 |
| RF EN 1 | 96 | 0.985 | 0.985 |
| RF EN 2 | 96 | 0.986 | 0.988 |
| RF GI 1 | 96 | 0.985 | 0.985 |
| RF GI 2 | 96 | 0.983 | 0.987 |
| Candidate AIM | 47 | 0.955 | 0.979 |
| SNP | Chr | Genomic Position (bp) | Gene(s) |
|---|---|---|---|
| AX-89926492 | 1 | 36,478,362 | ENSSTUG00000034565 |
| AX-89933844 | 3 | 56,808,067 | MTX1A, THBS3A, ENSSTUG00000008371, HJV, ITGA8, ENSSTUG00000009416 |
| AX-89957249 | 3 | 49,711,361 | ENSSTUG00000029747 |
| AX-89933361 | 4 | 43,428,599 | CBR4, SH3RF1 |
| AX-89954271 | 5 | 11,245,515 | GFRA1, CCDC172 |
| AX-89955512 | 5 | 31,053,287 | FUOM, ENSSTUG00000037533, ZGC:66426, ERLIN1 |
| AX-89964745 | 6 | 28,034,638 | MRC1A, SLC39A12, CACNB2 |
| AX-89965418 | 6 | 53,152,780 | USP9, ENSSTUG00000025824, DDX3XA, TGDS, GPR180, SLC5A3A, SI:CH211-132G1.7 |
| AX-89922103 | 8 | 11,821,548 | RGS12A, MSANTD1, DTX4A |
| AX-89923685 | 8 | 40,108,784 | ENSSTUG00000017894, ETV6, ENSSTUG00000017923 |
| AX-89930404 | 10 | 9,847,082 | FMNL1, ENSSTUG00000009061, GRB7 |
| AX-89926808 | 12 | 25,219,858 | CYP2R1, PDE3B |
| AX-89935881 | 12 | 68,862,681 | MED13L |
| AX-89941680 | 12 | 72,156,842 | GPSM1B, LHX3 |
| AX-89943019 | 12 | 78,807,143 | MPDU1A, ESRRA, KCNK4, STX5AL, EHD1B |
| AX-89944919 | 12 | 68,082,269 | ENSSTUG00000031577, TJP2A, ENSSTUG00000036079, SMC5, ZFAND5B |
| AX-89966227 | 12 | 24,351,329 | POLR2L, DAGLA, EXT2, SYT7B, SDHAF2, CPSF7 |
| AX-89937326 | 13 | 48,590,388 | GRIK4 |
| AX-89970985 | 13 | 27,301,563 | PPP3CB, UBE2D2, ENSSTUG00000035840, ENSSTUG00000035847, PSD2 |
| AX-89928338 | 14 | 30,569,008 | MRPL20, ATAD3A, TMEM240B, SSU72, ORA4, ENSSTUG00000008474, CCNL1B, VWA1 |
| AX-89965056 | 14 | 22,507,646 | GATA2A |
| AX-89976571 | 14 | 25,409,004 | FHIT |
| AX-89975434 | 15 | 23,622,069 | IFT46, VPS11, HYOU1, H2AX1, ZPR1 |
| AX-89971379 | 16 | 37,104,371 | PARK7, KCNAB2A |
| AX-89961240 | 19 | 43,548,455 | TMEM164, AMMECR1, KIF4, MRPS12, ENSSTUG00000016882, ENSSTUG00000016884, FIBPB |
| AX-89961754 | 21 | 24,970,403 | TMEM53, TESK2, TOE1 |
| AX-89969654 | 22 | 13,715,689 | MYO9B, S1PR4, MIR24-4, ENSSTUG00000015334, ENSSTUG00000015336 |
| AX-89957356 | 24 | 15,818,961 | DOCK9 |
| AX-89924719 | 25 | 31,489,217 | MYCLA, NT5C1AB, ENSSTUG00000048637 |
| AX-89935421 | 25 | 23,270,420 | ODC1, UTP25, ENSSTUG00000028825, ZGC:123321, LAMTOR3, ENSSTUG00000028866, ATP10D |
| AX-89950643 | 25 | 32,760,767 | RALGAPA2 |
| AX-89936803 | 26 | 26,628,471 | SRPRA, FAM118B, ILVBL, ENSSTUG00000048185, ENSSTUG00000048296, B3GAT1A |
| AX-89959464 | 26 | 22,256,971 | SLC47A4, SLC47A3, SLC13A5B, SERPINF2B, ENSSTUG00000049175, ENSSTUG00000049189, RPA1 |
| AX-89948079 | 27 | 20,927,232 | IGSF9B, ENSSTUG00000024232, TMEM127, CIAO1, SNRNP200, SLC20A1A |
| AX-89961304 | 28 | 42,593,898 | ENSSTUG00000021816, ASXL1, PCMTD2A, MYT1A |
| AX-89963552 | 28 | 24,003,249 | ENSSTUG00000043167, LRRC47, CEP104 |
| AX-89961685 | 29 | 17,322,240 | - |
| AX-89965310 | 33 | 12,906,283 | ENSSTUG00000029959 |
| AX-89927784 | 35 | 3,818,258 | FNDC1, OTOFA |
| AX-89958723 | 35 | 4,892,093 | ENSSTUG00000020096, TRMT6, FERMT1, BMP2B |
| AX-89938669 | 38 | 8,489,802 | RIMS1, KCNQ5B |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvatore, G.; Palombo, V.; Esposito, S.; Iaffaldano, N.; D’Andrea, M. Identification of Ancestry Informative Markers in Mediterranean Trout Populations of Molise (Italy): A Multi-Methodological Approach with Machine Learning. Genes 2022, 13, 1351. https://doi.org/10.3390/genes13081351
Salvatore G, Palombo V, Esposito S, Iaffaldano N, D’Andrea M. Identification of Ancestry Informative Markers in Mediterranean Trout Populations of Molise (Italy): A Multi-Methodological Approach with Machine Learning. Genes. 2022; 13(8):1351. https://doi.org/10.3390/genes13081351
Chicago/Turabian StyleSalvatore, Giovanna, Valentino Palombo, Stefano Esposito, Nicolaia Iaffaldano, and Mariasilvia D’Andrea. 2022. "Identification of Ancestry Informative Markers in Mediterranean Trout Populations of Molise (Italy): A Multi-Methodological Approach with Machine Learning" Genes 13, no. 8: 1351. https://doi.org/10.3390/genes13081351