
Gene Conversion Explains Elevated Diversity in the Immunity Modulating APL1 Gene of the Malaria Vector Anopheles funestus

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Identifying APL, LRIM, TEP and AMP Genes in An. funestus
2.2. Variant Prediction from Genome-Wide Pooled-Sequencing
2.3. Targeted Sequencing of Candidate Resistance Genes and Regions
2.4. Fst-Based Associations between Resistant and Susceptible Mosquitoes within Each Country
2.5. Gene Expression of APL1, TEP and LRIM Genes
2.6. Identifying Shared Polymorphisms and Recombination between APL1 Paralogues
3. Results
3.1. Immune Gene Complements of An. funestus
3.2. APL1 Has Elevated Diversity
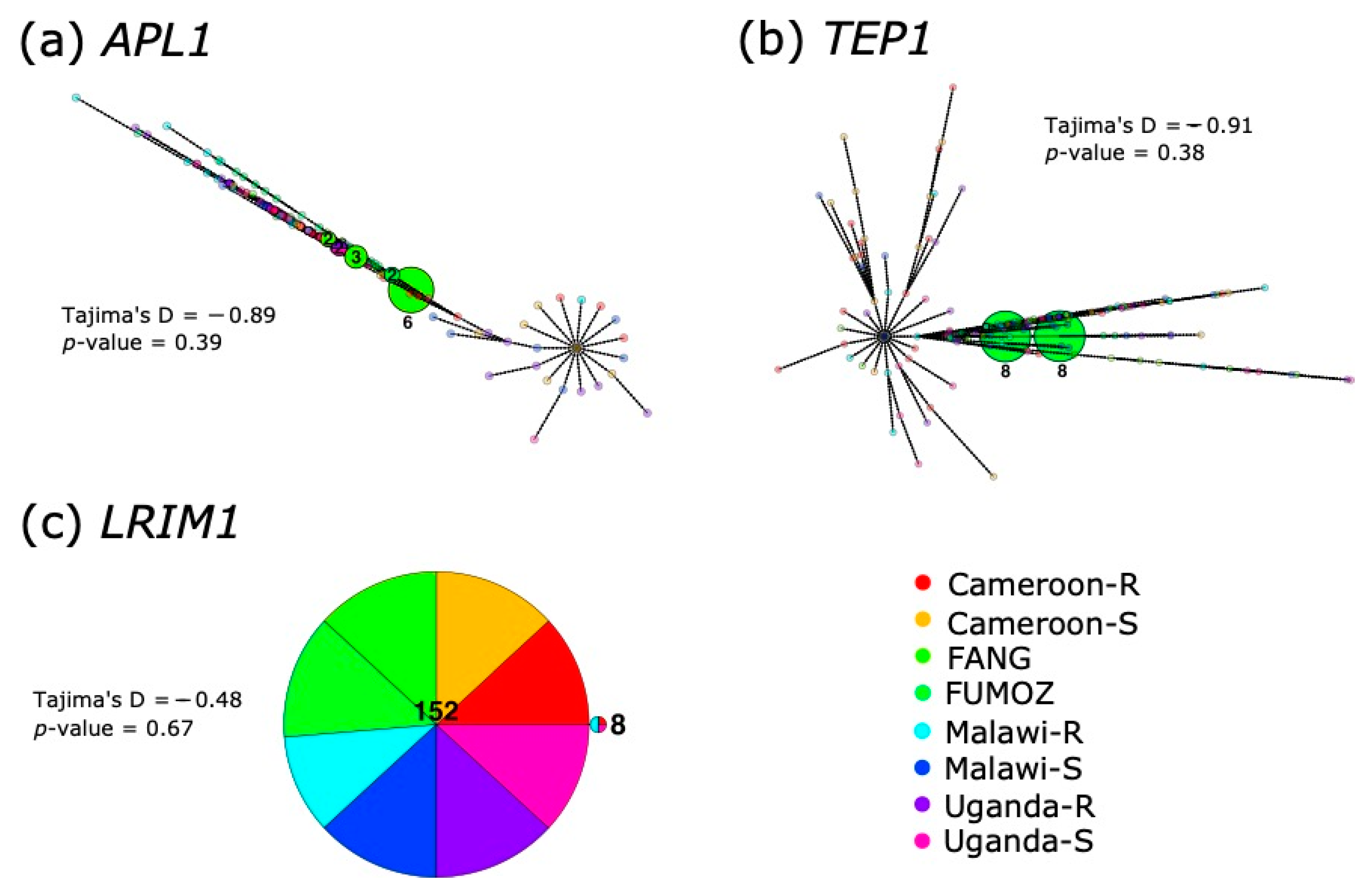
3.3. Haplotype Analyses and Fst between Countries Reveals No Clustering by Resistance or Origin
3.4. Gambicin and Other Immune-Related Genes Are Also Highly Polymorphic
3.5. APL1 Expression Is Greater Than Its Paralogues and a Subset of AMPs Are Very Highly Expressed
3.6. Discordant Read Mappings Consistent with Gene Conversion between APL1 Paralogues
4. Discussion
4.1. Gene Conversion Explains Elevated APL1 Diversity in An. funestus
4.2. Gene Conversion Acts on APL1 Orthologues in Other Anopheles Species
4.3. Elevated πN in the An. funestus Anti-Microbial Protein Gambicin
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. World Malaria Report 2020: 20 Years of Global Progress and Challenges; World Malaria Report: Geneva, Switzerland, 2020; pp. 1–151. [Google Scholar]
- White, B.J.; Lawniczak, M.K.N.; Cheng, C.; Coulibaly, M.B.; Wilson, M.D.; Sagnon, N.; Costantini, C.; Simard, F.; Christophides, G.K.; Besansky, N.J. Adaptive divergence between incipient species of Anopheles gambiae increases resistance to Plasmodium. Proc. Natl. Acad. Sci. USA 2011, 108, 244–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blandin, S.A.; Wang-Sattler, R.; Lamacchia, M.; Gagneur, J.; Lycett, G.; Ning, Y.; Levashina, E.A.; Steinmetz, L.M. Dissecting the genetic basis of resistance to malaria parasites in Anopheles gambiae. Science 2009, 326, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Rivero, A.; Vezilier, J.; Weill, M.; Read, A.F.; Gandon, S. Insecticide control of vector-borne diseases: When is insecticide resistance a problem? PLoS Pathog. 2010, 6, e1001000. [Google Scholar] [CrossRef] [Green Version]
- Clayton, A.M.; Dong, Y.; Dimopoulos, G. The Anopheles innate immune system in the defense against malaria infection. J. Innate Immun. 2014, 6, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.H.G.; Chang, C.-I.; Chelliah, Y.; Blandin, S.; Levashina, E.A.; Deisenhofer, J. Structural basis for conserved complement factor-like function in the antimalarial protein TEP1. Proc. Natl. Acad. Sci. USA 2007, 104, 11615–11620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povelones, M.; Bhagavatula, L.; Yassine, H.; Tan, L.A.; Upton, L.M.; Osta, M.A.; Christophides, G.K. The CLIP-domain serine protease homolog SPCLIP1 regulates complement recruitment to microbial surfaces in the malaria mosquito Anopheles gambiae. PLoS Pathog. 2013, 9, e1003623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes Ruiz, V.M.; Sousa, G.L.; Sneed, S.D.; Farrant, K.V.; Christophides, G.K.; Povelones, M. Stimulation of a protease targeting the LRIM1/APL1C complex reveals specificity in complement-like pathway activation in Anopheles gambiae. PLoS ONE 2019, 14, e0214753. [Google Scholar] [CrossRef]
- Wu, Q.; Patočka, J.; Kuča, K. Insect antimicrobial peptides, a mini review. Toxins 2018, 10, 461. [Google Scholar] [CrossRef]
- Lee, W.-S.; Webster, J.A.; Madzokere, E.T.; Stephenson, E.B.; Herrero, L.J. Mosquito antiviral defense mechanisms: A delicate balance between innate immunity and persistent viral infection. Parasit. Vectors 2019, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Vizioli, J.; Bulet, P.; Hoffmann, J.A.; Kafatos, F.C.; Müller, H.-M.; Dimopoulos, G. Gambicin: A novel immune responsive antimicrobial peptide from the malaria vector Anopheles gambiae. Proc. Natl. Acad. Sci. USA 2001, 98, 12630–12635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Cruz, A.; Garver, L.S.; Alabaster, A.; Bangiolo, L.; Haile, A.; Winikor, J.; Ortega, C.; van Schaijk, B.C.L.; Sauerwein, R.W.; Taylor-Salmon, E.; et al. The human malaria parasite Pfs47 gene mediates evasion of the mosquito immune system. Science 2013, 340, 984–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, S.-J.; Jacobs-Lorena, M. A Plasmodium key fits a mosquito lock. Proc. Natl. Acad. Sci. USA 2020, 117, 3898–3900. [Google Scholar] [CrossRef] [PubMed]
- Molina-Cruz, A.; DeJong, R.J.; Ortega, C.; Haile, A.; Abban, E.; Rodrigues, J.; Jaramillo-Gutierrez, G.; Barillas-Mury, C. Some strains of Plasmodium falciparum, a human malaria parasite, evade the complement-like system of Anopheles gambiae mosquitoes. Proc. Natl. Acad. Sci. USA 2012, 109, E1957–E1962. [Google Scholar] [CrossRef] [Green Version]
- Povelones, M.; Waterhouse, R.M.; Kafatos, F.C.; Christophides, G.K. Leucine-rich repeat protein complex activates mosquito complement in defense against Plasmodium parasites. Science 2009, 324, 258–261. [Google Scholar] [CrossRef] [Green Version]
- Niaré, O.; Markianos, K.; Volz, J.; Oduol, F.; Touré, A.; Bagayoko, M.; Sangaré, D.; Traoré, S.F.; Wang, R.; Blass, C.; et al. Genetic loci affecting resistance to human malaria parasites in a West African mosquito vector population. Science 2002, 298, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Riehle, M.M.; Markianos, K.; Niaré, O.; Xu, J.; Li, J.; Touré, A.M.; Podiougou, B.; Oduol, F.; Diawara, S.; Diallo, M.; et al. Natural malaria infection in Anopheles gambiae is regulated by a single genomic control region. Science 2006, 312, 577–579. [Google Scholar] [CrossRef] [Green Version]
- Rottschaefer, S.M.; Riehle, M.M.; Coulibaly, B.; Sacko, M.; Niare, O.; Morlais, I.; Traore, S.F.; Vernick, K.D.; Lazzaro, B.P. Exceptional diversity, maintenance of polymorphism, and recent directional selection on the APL1 malaria resistance genes of Anopheles gambiae. PLoS Biol. 2011, 9, e1000600. [Google Scholar] [CrossRef] [Green Version]
- Obbard, D.J.; Callister, D.M.; Jiggins, F.M.; Soares, D.C.; Yan, G.; Little, T.J. The evolution of TEP1, an exceptionally polymorphic immunity gene in Anopheles gambiae. BMC Evol. Biol. 2008, 8, 274. [Google Scholar] [CrossRef] [Green Version]
- Mitri, C.; Bischoff, E.; Eiglmeier, K.; Holm, I.; Dieme, C.; Brito-Fravallo, E.; Raz, A.; Zakeri, S.; Nejad, M.I.K.; Djadid, N.D.; et al. Gene copy number and function of the APL1 immune factor changed during Anopheles evolution. Parasit. Vectors 2020, 13, 18. [Google Scholar] [CrossRef] [Green Version]
- Mitri, C.; Bischoff, E.; Belda Cuesta, E.; Volant, S.; Ghozlane, A.; Eiglmeier, K.; Holm, I.; Dieme, C.; Brito-Fravallo, E.; Guelbeogo, W.M.; et al. Leucine-Rich immune factor APL1 is associated with specific modulation of enteric microbiome taxa in the asian malaria mosquito Anopheles stephensi. Front. Microbiol. 2020, 11, 306. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.R.; Hill, T.; Unckless, R.L. Balancing Selection Drives the Maintenance of Genetic Variation in Drosophila Antimicrobial Peptides. Genome Biol. Evol. 2019, 11, 2691–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, T.; Hume, J.C.C.; Licht, M.; Burns, C.S.; Wollenberg, K.; Simard, F.; Ribeiro, J. Molecular evolution of immune genes in the malaria mosquito Anopheles gambiae. PLoS ONE 2009, 4, e4549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slotman, M.A.; Parmakelis, A.; Marshall, J.C.; Awono-Ambene, P.H.; Antonio-Nkondjo, C.; Simard, F.; Caccone, A.; Powell, J.R. Patterns of selection in anti-malarial immune genes in malaria vectors: Evidence for adaptive evolution in LRIM1 in Anopheles arabiensis. PLoS ONE 2007, 2, e793. [Google Scholar] [CrossRef]
- Gillies, M.T.; De Meillon, B. The Anophelinae of Africa South of the Sahara (Ethiopian Zoogeographical Region); South African Institute for Medical Research: Johannesburg, South Africa, 1968; Volume 54. [Google Scholar]
- Coetzee, M.; Fontenille, D. Advances in the study of Anopheles funestus, a major vector of malaria in Africa. Insect Biochem. Mol. Biol. 2004, 34, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.C.; Irving, H.; Okedi, L.M.; Steven, A.; Wondji, C.S. Pyrethroid resistance in an Anopheles funestus population from Uganda. PLoS ONE 2010, 5, e11872. [Google Scholar] [CrossRef]
- Ghurye, J.; Koren, S.; Small, S.T.; Redmond, S.; Howell, P.; Phillippy, A.M.; Besansky, N.J. A chromosome-scale assembly of the major African malaria vector Anopheles funestus. Gigascience 2019, 8, giz063. [Google Scholar] [CrossRef]
- Weedall, G.D.; Mugenzi, L.M.J.; Menze, B.D.; Tchouakui, M.; Ibrahim, S.S.; Amvongo-Adjia, N.; Irving, H.; Wondji, M.J.; Tchoupo, M.; Djouaka, R.; et al. A cytochrome P450 allele confers pyrethroid resistance on a major African malaria vector, reducing insecticide-treated bednet efficacy. Sci. Transl. Med. 2019, 11, eaat7386. [Google Scholar] [CrossRef] [Green Version]
- Riveron, J.M.; Yunta, C.; Ibrahim, S.S.; Djouaka, R.; Irving, H.; Menze, B.D.; Ismail, H.M.; Hemingway, J.; Ranson, H.; Albert, A.; et al. A single mutation in the GSTe2 gene allows tracking of metabolically based insecticide resistance in a major malaria vector. Genome Biol. 2014, 15, R27. [Google Scholar] [CrossRef] [Green Version]
- Mugenzi, L.M.J.; Menze, B.D.; Tchouakui, M.; Wondji, M.J.; Irving, H.; Tchoupo, M.; Hearn, J.; Weedall, G.D.; Riveron, J.M.; Wondji, C.S. Cis-regulatory CYP6P9b P450 variants associated with loss of insecticide-treated bed net efficacy against Anopheles funestus. Nat. Commun. 2019, 10, 4652. [Google Scholar] [CrossRef] [Green Version]
- Tchouakui, M.; Chiang, M.-C.; Ndo, C.; Kuicheu, C.K.; Amvongo-Adjia, N.; Wondji, M.J.; Tchoupo, M.; Kusimo, M.O.; Riveron, J.M.; Wondji, C.S. A marker of glutathione S-transferase-mediated resistance to insecticides is associated with higher Plasmodium infection in the African malaria vector Anopheles funestus. Sci. Rep. 2019, 9, 5772. [Google Scholar] [CrossRef] [Green Version]
- Viljakainen, L. Evolutionary genetics of insect innate immunity. Brief. Funct. Genom. 2015, 14, 407–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sackton, T.B.; Lazzaro, B.P.; Clark, A.G. Rapid expansion of immune-related gene families in the house fly, Musca domestica. Mol. Biol. Evol. 2017, 34, 857–872. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weedall, G.D.; Riveron, J.M.; Hearn, J.; Irving, H.; Kamdem, C.; Fouet, C.; White, B.J.; Wondji, C.S. An Africa-wide genomic evolution of insecticide resistance in the malaria vector Anopheles funestus involves selective sweeps, copy number variations, gene conversion and transposons. PLOS Genet. 2020, 16, e1008822. [Google Scholar] [CrossRef]
- Hearn, J.; Djoko Tagne, C.S.; Ibrahim, S.S.; Tene-Fossog, B.; Mugenzi, L.M.J.; Irving, H.; Riveron, J.M.; Weedall, G.D.; Wondji, C.S. Multi-omics analysis identifies a CYP9K1 haplotype conferring pyrethroid resistance in the malaria vector Anopheles funestus in East Africa. Mol. Ecol. 2022. accepted. [Google Scholar] [CrossRef] [PubMed]
- Picard Toolkit. Broad Institute, GitHub Repos. 2019. Available online: https://github.com/broadinstitute (accessed on 19 May 2022).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.W.; Moncla, L.H.; Hughes, A.L. SNPGenie: Estimating evolutionary parameters to detect natural selection using pooled next-generation sequencing data. Bioinformatics 2015, 31, 3709–3711. [Google Scholar] [CrossRef] [Green Version]
- Hunt, R.H.; Brooke, B.D.; Pillay, C.; Koekemoer, L.L.; Coetzee, M. Laboratory selection for and characteristics of pyrethroid resistance in the malaria vector Anopheles funestus. Med. Vet. Entomol. 2005, 19, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Wondji, C.S.; Irving, H.; Morgan, J.; Lobo, N.F.; Collins, F.H.; Hunt, R.H.; Coetzee, M.; Hemingway, J.; Ranson, H. Two duplicated P450 genes are associated with pyrethroid resistance in Anopheles funestus, a major malaria vector. Genome Res. 2009, 19, 452–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar] [CrossRef]
- Patterson, M.; Marschall, T.; Pisanti, N.; Van Iersel, L.; Stougie, L.; Klau, G.W.; Schönhuth, A. WhatsHap: Weighted haplotype assembly for future-generation sequencing reads. J. Comput. Biol. 2015, 22, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Pertea, M. GFF utilities: GffRead and GffCompare [version 2; peer review: 3 approved]. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E. pegas: An R package for population genetics with an integrated—Modular approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef] [Green Version]
- Hivert, V.; Leblois, R.; Petit, E.J.; Gautier, M.; Vitalis, R. Measuring genetic differentiation from Pool-seq data. Genetics 2018, 210, 315–330. [Google Scholar] [CrossRef] [Green Version]
- Dabney, A.; Storey, J.D.; Warnes, G.R. Qvalue: Q-Value Estimation for False Discovery Rate Control; R Packag. Version 2.22.0. 2021. p. 1. Available online: https://www.bioconductor.org/packages/devel/bioc/manuals/qvalue/man/qvalue.pdf (accessed on 19 May 2022).
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 534. [Google Scholar] [CrossRef] [Green Version]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwiczak, J.; Winski, A.; Szczepaniak, K.; Alva, V.; Dunin-Horkawicz, S. DeepCoil—a fast and accurate prediction of coiled-coil domains in protein sequences. Bioinformatics 2019, 35, 2790–2795. [Google Scholar] [CrossRef] [PubMed]
- Neafsey, D.E.; Waterhouse, R.M.; Abai, M.R.; Aganezov, S.S.; Alekseyev, M.A.; Allen, J.E.; Amon, J.; Arcà, B.; Arensburger, P.; Artemov, G.; et al. Highly evolvable malaria vectors: The genomes of 16 Anopheles mosquitoes. Science 2015, 347, 1258522. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurosawa, K.; Ohta, K. Genetic diversification by somatic gene conversion. Genes 2011, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, B.I.; Barbour, A.G. Antigen diversity in the bacterium B. hermsii through “somatic” mutations in rearranged vmp genes. Cell 1994, 78, 867–876. [Google Scholar] [CrossRef]
- Morrison, L.J.; Marcello, L.; McCulloch, R. Antigenic variation in the African trypanosome: Molecular mechanisms and phenotypic complexity. Cell Microbiol. 2009, 11, 1724–1734. [Google Scholar] [CrossRef] [Green Version]
- Unckless, R.L.; Lazzaro, B.P. The potential for adaptive maintenance of diversity in insect antimicrobial peptides. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150291. [Google Scholar] [CrossRef] [Green Version]
- Arcà, B.; Struchiner, C.J.; Pham, V.M.; Sferra, G.; Lombardo, F.; Pombi, M.; Ribeiro, J.M.C. Positive selection drives accelerated evolution of mosquito salivary genes associated with blood-feeding. Insect Mol. Biol. 2014, 23, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Lazzaro, B.P. Natural selection on the Drosophila antimicrobial immune system. Curr. Opin. Microbiol. 2008, 11, 284–289. [Google Scholar] [CrossRef] [Green Version]
- Sackton, T.B.; Lazzaro, B.P.; Schlenke, T.A.; Evans, J.D.; Hultmark, D.; Clark, A.G. Dynamic evolution of the innate immune system in Drosophila. Nat. Genet. 2007, 39, 1461–1468. [Google Scholar] [CrossRef] [PubMed]
- Brady, D.; Grapputo, A.; Romoli, O.; Sandrelli, F. Insect cecropins, antimicrobial peptides with potential therapeutic applications. Int. J. Mol. Sci. 2019, 20, 5862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchouakui, M.; Miranda, J.R.; Mugenzi, L.M.J.; Djonabaye, D.; Wondji, M.J.; Tchoupo, M.; Tchapga, W.; Njiokou, F.; Wondji, C.S. Cytochrome P450 metabolic resistance (CYP6P9a) to pyrethroids imposes a fitness cost in the major African malaria vector Anopheles funestus. Heredity 2020, 124, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Tchouakui, M.; Mugenzi, L.M.J.; Wondji, M.J.; Tchoupo, M.; Njiokou, F.; Wondji, C.S. Combined over-expression of two cytochrome P450 genes exacerbates the fitness cost of pyrethroid resistance in the major African malaria vector Anopheles funestus. Pestic. Biochem. Physiol. 2021, 173, 104772. [Google Scholar] [CrossRef]
- Freitak, D.; Wheat, C.W.; Heckel, D.G.; Vogel, H. Immune system responses and fitness costs associated with consumption of bacteria in larvae of Trichoplusia ni. BMC Biol. 2007, 5, 56. [Google Scholar] [CrossRef] [Green Version]
- Fellous, S.; Lazzaro, B.P. Potential for evolutionary coupling and decoupling of larval and adult immune gene expression. Mol. Ecol. 2011, 20, 1558–1567. [Google Scholar] [CrossRef] [Green Version]
- Kouamo, M.F.M.; Ibrahim, S.S.; Hearn, J.; Riveron, J.M.; Kusimo, M.; Tchouakui, M.; Ebai, T.; Tchapga, W.; Wondji, M.J.; Irving, H.; et al. Genome-wide transcriptional analysis and functional validation linked a cluster of epsilon glutathione S-transferases with insecticide resistance in the major malaria vector Anopheles funestus across Africa. Genes 2021, 12, 561. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Nonsynonymous Sites | Synonymous Sites | πN | πS |
|---|---|---|---|---|
| PoolSeq | ||||
| APL1/AFUN018743 | 1475.80 | 426.20 | 0.027 | 0.036 |
| AFUN018581 | 1396.35 | 409.62 | 0.023 | 0.036 |
| AFUN000279 | 1399.86 | 406.14 | 0.010 | 0.012 |
| AFUN000288 | 2320.70 | 688.30 | 0.012 | 0.019 |
| AFUN000597 | 1401.36 | 404.64 | 0.008 | 0.008 |
| SureSelect | ||||
| APL1/AFUN018743 | 1461.21 | 418.62 | 0.025 | 0.027 |
| AFUN018581 | 1389.97 | 402.91 | 0.019 | 0.023 |
| AFUN000279 | 1389.10 | 402.13 | 0.022 | 0.028 |
| AFUN000288 | 2313.82 | 689.23 | 0.017 | 0.025 |
| AFUN000597 | 1398.40 | 404.38 | 0.018 | 0.020 |
| Gene | Original Coverage | Discordant Read Filtered Coverage |
|---|---|---|
| AFUN018743 | 48.31 | 42.79 |
| AFUN018581 | 49.55 | 43.16 |
| AFUN000279 | 20.79 | 17.06 |
| AFUN000288 | 23.12 | 20.48 |
| AFUN000597 | 14.60 | 12.15 |
| All genes | 33.98 | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hearn, J.; Riveron, J.M.; Irving, H.; Weedall, G.D.; Wondji, C.S. Gene Conversion Explains Elevated Diversity in the Immunity Modulating APL1 Gene of the Malaria Vector Anopheles funestus. Genes 2022, 13, 1102. https://doi.org/10.3390/genes13061102
Hearn J, Riveron JM, Irving H, Weedall GD, Wondji CS. Gene Conversion Explains Elevated Diversity in the Immunity Modulating APL1 Gene of the Malaria Vector Anopheles funestus. Genes. 2022; 13(6):1102. https://doi.org/10.3390/genes13061102
Chicago/Turabian StyleHearn, Jack, Jacob M. Riveron, Helen Irving, Gareth D. Weedall, and Charles S. Wondji. 2022. "Gene Conversion Explains Elevated Diversity in the Immunity Modulating APL1 Gene of the Malaria Vector Anopheles funestus" Genes 13, no. 6: 1102. https://doi.org/10.3390/genes13061102