
Integrated Analysis of Tissue-Specific Gene Expression in Diabetes by Tensor Decomposition Can Identify Possible Associated Diseases



Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression
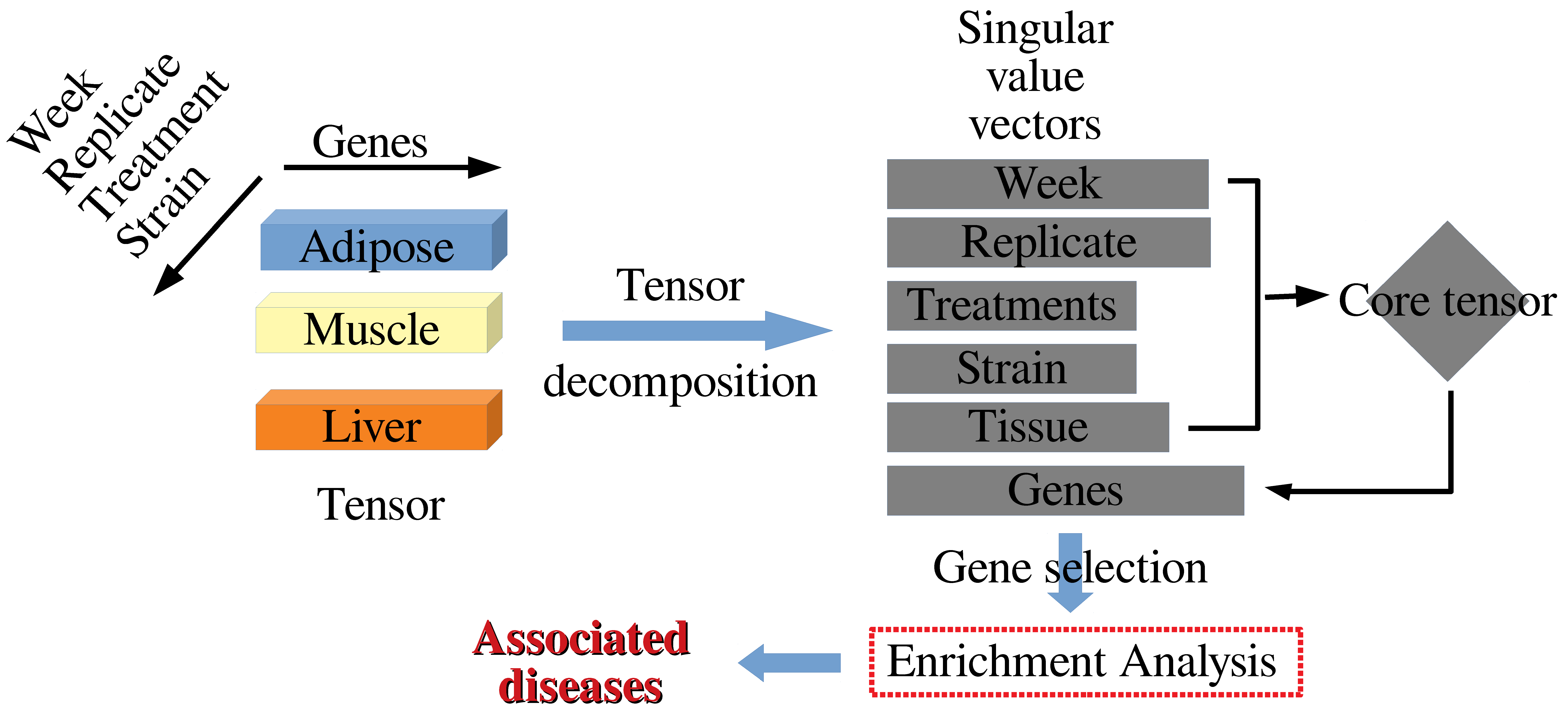
2.2. Methods
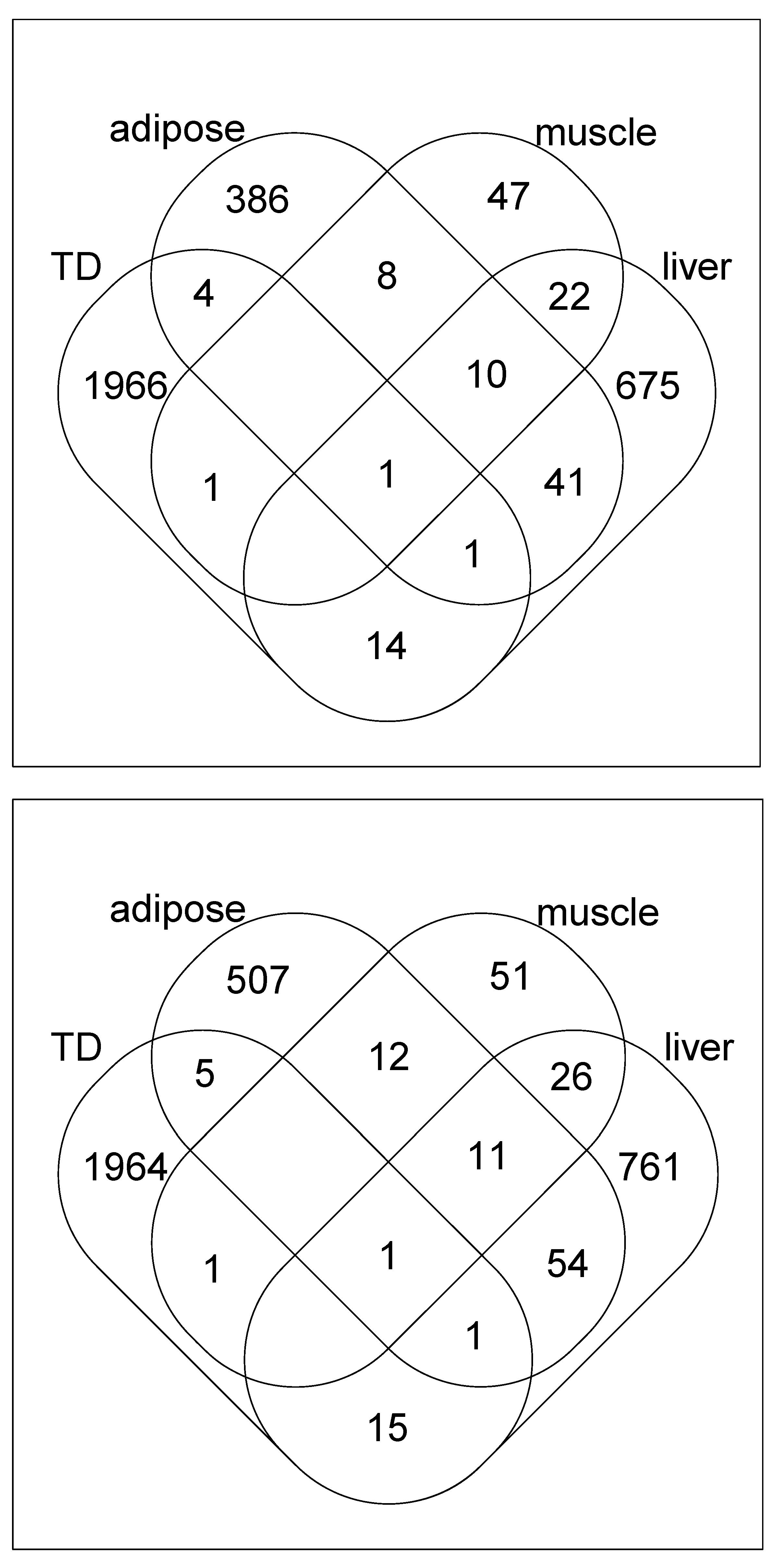
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taguchi, Y.H. Unsupervised Feature Extraction Applied to Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar] [CrossRef]
- Taguchi, Y.H.; Turki, T. Tensor decomposition- and principal component analysis-based unsupervised feature extraction to select more reasonable differentially expressed genes: Optimization of standard deviation versus state-of-art methods. bioRxiv 2022. [Google Scholar] [CrossRef]
- Taguchi, Y.H.; Turki, T. Principal component analysis- and tensor decomposition-based unsupervised feature extraction to select more reasonable differentially methylated cytosines: Optimization of standard deviation versus state-of-the-art methods. bioRxiv 2022. [Google Scholar] [CrossRef]
- Roy, S.S.; Taguchi, Y.H. Tensor decomposition and principal component analysis-based unsupervised feature extraction outperforms state-of-the-art methods when applied to histone modification profiles. bioRxiv 2022. [Google Scholar]
- Xue, B.; Nie, J.; Wang, X.; DuBois, D.C.; Jusko, W.J.; Almon, R.R. Effects of High Fat Feeding on Adipose Tissue Gene Expression in Diabetic Goto-Kakizaki Rats. Gene Regul. Syst. Biol. 2015, 9, GRSB.S25172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Umegaki, H. Neurodegeneration in Diabetes Mellitus. In Neurodegenerative Diseases; Ahmad, S.I., Ed.; Springer: New York, NY, USA, 2012; pp. 258–265. [Google Scholar] [CrossRef]
- Ristow, M. Neurodegenerative disorders associated with diabetes mellitus. J. Mol. Med. 2004, 82, 510–529. [Google Scholar] [CrossRef]
- Nasrolahi, A.; Mahmoudi, J.; Noori-Zadeh, A.; Haghani, K.; Bakhtiyari, S.; Darabi, S. Shared Pathological Mechanisms Between Diabetes Mellitus and Neurodegenerative Diseases. Curr. Pharmacol. Rep. 2019, 5, 219–231. [Google Scholar] [CrossRef]
- Madhusudhanan, J.; Suresh, G.; Devanathan, V. Neurodegeneration in type 2 diabetes: Alzheimer’s as a case study. Brain Behav. 2020, 10, e01577. [Google Scholar] [CrossRef] [Green Version]
- León, K.I.L.D.; Bertadillo-Jilote, A.D.; García-Gutiérrez, D.G.; Meraz-Ríos, M.A. Alzheimer’s Disease and Type 2 Diabetes Mellitus: Molecular Mechanisms and Similarities. In Neurodegenerative Diseases; Tunalı, N.E., Ed.; IntechOpen: Rijeka, Croatia, 2020; Chapter 4. [Google Scholar] [CrossRef]
- Sun, H.; Wang, Y. A new branch connecting thermogenesis and diabetes. Nat. Metab. 2019, 1, 845–846. [Google Scholar] [CrossRef]
- Lewis, M.T.; Kasper, J.D.; Bazil, J.N.; Frisbee, J.C.; Wiseman, R.W. Quantification of Mitochondrial Oxidative Phosphorylation in Metabolic Disease: Application to Type 2 Diabetes. Int. J. Mol. Sci. 2019, 20, 5271. [Google Scholar] [CrossRef] [Green Version]
- Holm, L.J.; Mnsted, M.O.; Haupt-Jorgensen, M.; Buschard, K. PPARs and the Development of Type 1 Diabetes. PPAR Res. 2020, 2020, 6198628. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2006, 8, 118–127. [Google Scholar] [CrossRef]
- Taguchi, Y.H.; Wang, H. Genetic Association between Amyotrophic Lateral Sclerosis and Cancer. Genes 2017, 8, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, Y.H.; Iwadate, M.; Umeyama, H. Principal component analysis-based unsupervised feature extraction applied to in silico drug discovery for posttraumatic stress disorder-mediated heart disease. BMC Bioinform. 2015, 16, 139. [Google Scholar] [CrossRef] [Green Version]
- Babbi, G.; Martelli, P.L.; Profiti, G.; Bovo, S.; Savojardo, C.; Casadio, R. eDGAR: A database of Disease-Gene Associations with annotated Relationships among genes. BMC Genom. 2017, 18, 554. [Google Scholar] [CrossRef]
- Luo, P.; Xiao, Q.; Wei, P.J.; Liao, B.; Wu, F.X. Identifying Disease-Gene Associations With Graph-Regularized Manifold Learning. Front. Genet. 2019, 10, 270. [Google Scholar] [CrossRef]
- Opap, K.; Mulder, N. Recent advances in predicting gene–disease associations [version 1; peer review: 2 approved]. F1000Research 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Qiu, Y.; Li, Q.; Liu, S.; Ni, F. Predicting Drug-Disease Associations via Multi-Task Learning Based on Collective Matrix Factorization. Front. Bioeng. Biotechnol. 2020, 8, 218. [Google Scholar] [CrossRef]
- Jiang, H.; Huang, Y. An effective drug-disease associations prediction model based on graphic representation learning over multi-biomolecular network. BMC Bioinform. 2022, 23, 9. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Huang, F.; Zhao, X.; Xiao, W.; Zhang, W. Predicting drug–disease associations through layer attention graph convolutional network. Briefings Bioinform. 2020, 22, bbaa243. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Term | Overlap | p-Value | Adjusted p-Value |
|---|---|---|---|
| Diabetic cardiomyopathy | 83/203 | ||
| Prion disease | 93/273 | ||
| Parkinson disease | 86/249 | ||
| Oxidative phosphorylation | 60/133 | ||
| Nonalcoholic fatty liver disease | 65/155 | ||
| Thermogenesis | 76/232 | ||
| Complement and coagulation cascades | 42/85 | ||
| PPAR signaling pathway | 39/74 | ||
| Alzheimer disease | 94/369 | ||
| Huntington disease | 83/306 |
| Term | Overlap | p-Value | Adjusted p-Value |
|---|---|---|---|
| LIVER (BULK TISSUE) | 481/2316 | ||
| VENTRICLE | 449/2316 | ||
| SKELETAL MUSCLE (BULK TISSUE) | 428/2316 | ||
| ADIPOSE (BULK TISSUE) | 410/2316 | ||
| MYOBLAST | 409/2316 | ||
| SUBCUTANEOUS ADIPOSE TISSUE | 401/2316 | ||
| ATRIUM | 366/2316 | ||
| HEART (BULK TISSUE) | 363/2316 | ||
| HEPATOCYTE | 362/2316 | ||
| OMENTUM | 350/2316 |
| Term | Overlap | p-Value | Adjusted p-Value |
|---|---|---|---|
| mammary gland non-lactating | 116/201 | ||
| skeletal muscle | 229/710 | ||
| liver | 243/928 | ||
| adipose brown | 148/456 | ||
| heart | 154/568 | ||
| kidney | 80/554 | ||
| osteoblast day 21 | 44/264 | ||
| bladder | 33/195 | ||
| adipose white | 33/199 | ||
| MEF | 45/300 |
| Tissue | t Test | Sam | Limma |
|---|---|---|---|
| Adipose | 556 | 773 | 116 |
| Muscle | 100 | 119 | 2 |
| liver | 947 | 1090 | 211 |
| ComBat | 4009 | 180 | 0 |
| Term | Overlap | p-Value | Adjusted p-Value |
|---|---|---|---|
| t test | |||
| Adipose | |||
| adipose brown | 38/456 | ||
| mammary gland lact | 12/104 | ||
| macrophage peri LPS thio 0 h | 18/353 | ||
| Muscle | |||
| adipose brown | 29/456 | ||
| heart | 21/568 | ||
| mammary gland lact | 4/104 | ||
| Liver | |||
| liver | 90/928 | ||
| adipose brown | 40/456 | ||
| kidney | 40/554 | ||
| ComBat | |||
| bone marrow | 107/413 | ||
| osteoblast day 21 | 75/264 | ||
| embryonic stem line V26 2 p16 | 149/728 | ||
| sam | |||
| Adipose | |||
| adipose brown | 51/456 | ||
| mammary gland lact | 12/104 | ||
| macrophage peri LPS thio 0 h | 23/353 | ||
| Muscle | |||
| adipose brown | 33/456 | ||
| heart | 23/568 | ||
| mammary gland lact | 4/104 | ||
| Liver | |||
| liver | 93/928 | ||
| adipose brown | 43/456 | ||
| kidney | 43/554 | ||
| Cell cycle | 11/124 | ||
| Oocyte meiosis | 9/129 | ||
| Progesterone-mediated oocyte maturation | 8/100 | ||
| limma | |||
| Adipose | |||
| adipose brown | 14/456 | ||
| adipose white | 4/199 | ||
| intestine small | 6/466 | ||
| Liver | |||
| liver | 33/928 | ||
| adipose brown | 7/456 | ||
| heart | 8/568 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taguchi, Y.-H.; Turki, T. Integrated Analysis of Tissue-Specific Gene Expression in Diabetes by Tensor Decomposition Can Identify Possible Associated Diseases. Genes 2022, 13, 1097. https://doi.org/10.3390/genes13061097
Taguchi Y-H, Turki T. Integrated Analysis of Tissue-Specific Gene Expression in Diabetes by Tensor Decomposition Can Identify Possible Associated Diseases. Genes. 2022; 13(6):1097. https://doi.org/10.3390/genes13061097
Chicago/Turabian StyleTaguchi, Y-H., and Turki Turki. 2022. "Integrated Analysis of Tissue-Specific Gene Expression in Diabetes by Tensor Decomposition Can Identify Possible Associated Diseases" Genes 13, no. 6: 1097. https://doi.org/10.3390/genes13061097