
MDSCMF: Matrix Decomposition and Similarity-Constrained Matrix Factorization for miRNA–Disease Association Prediction


Abstract
:1. Introduction
2. Results
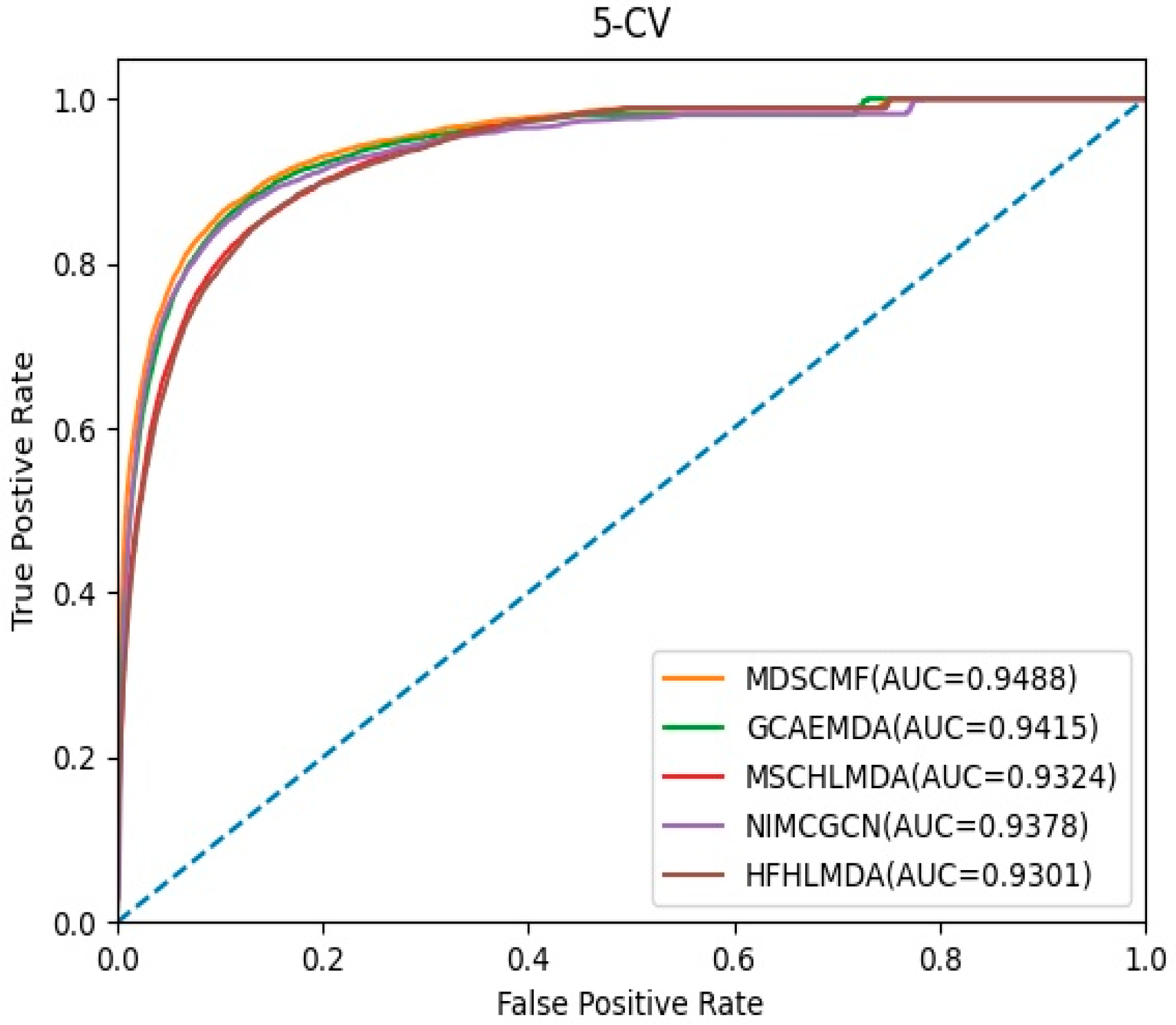
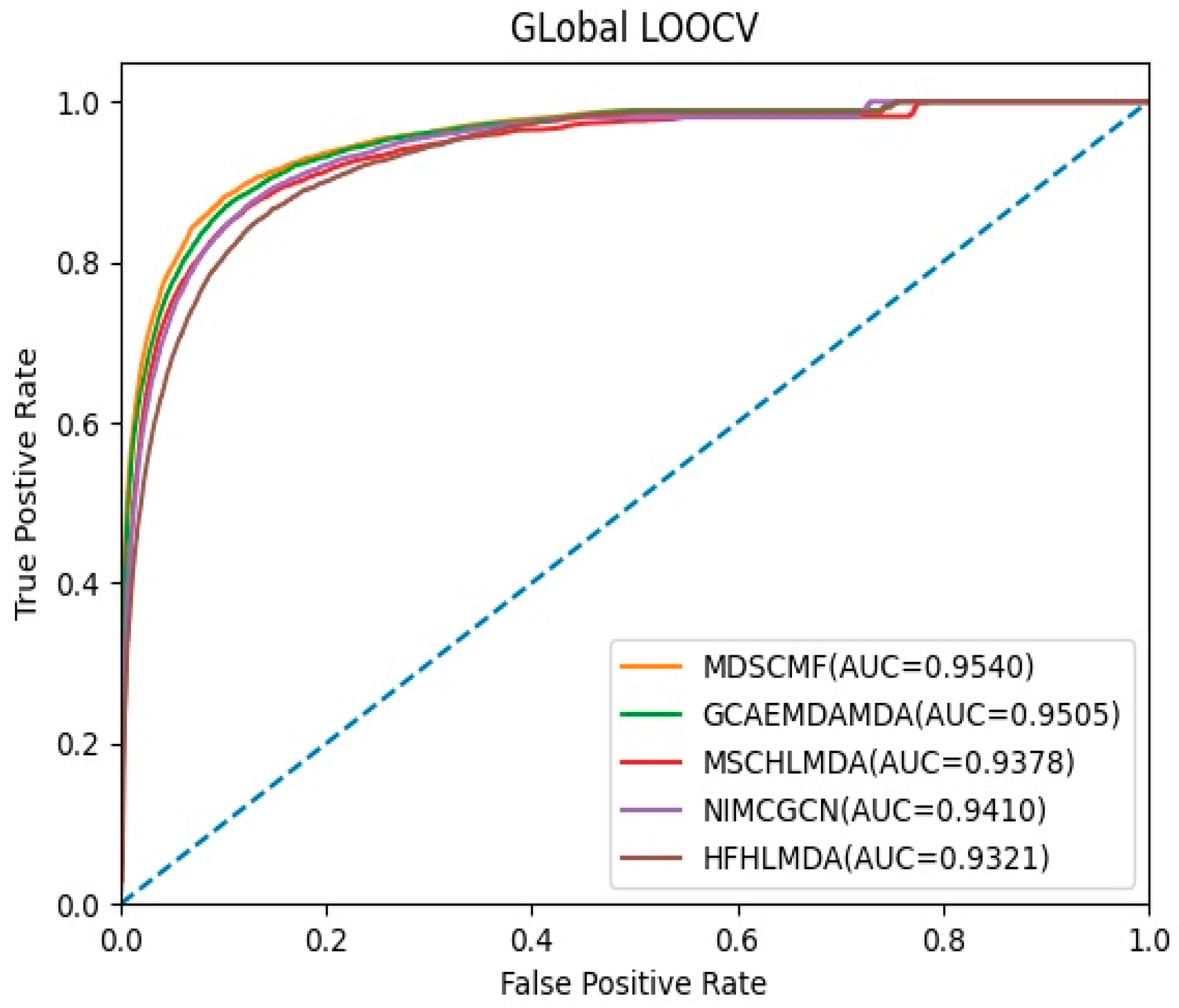
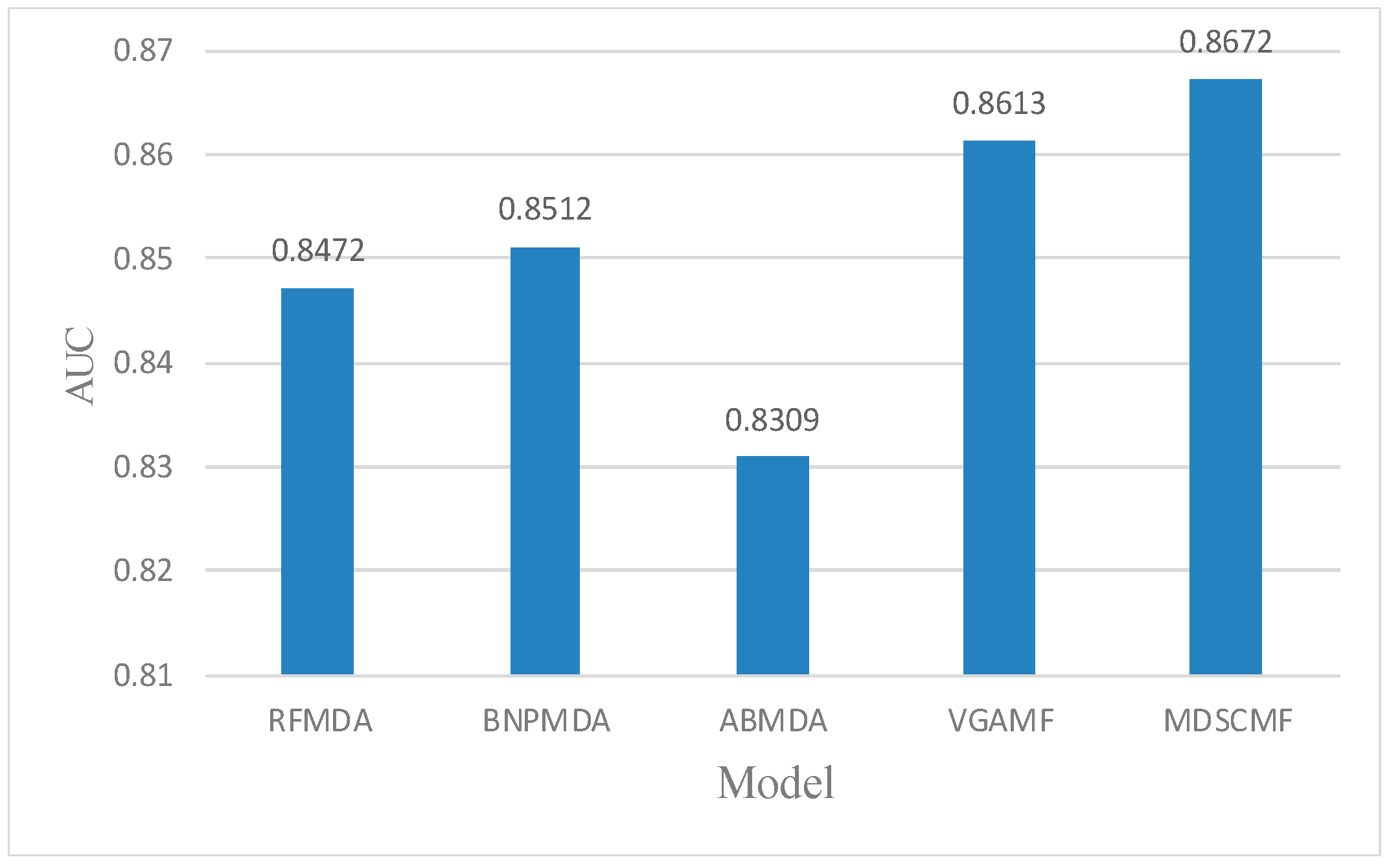
2.1. Performance Evaluation
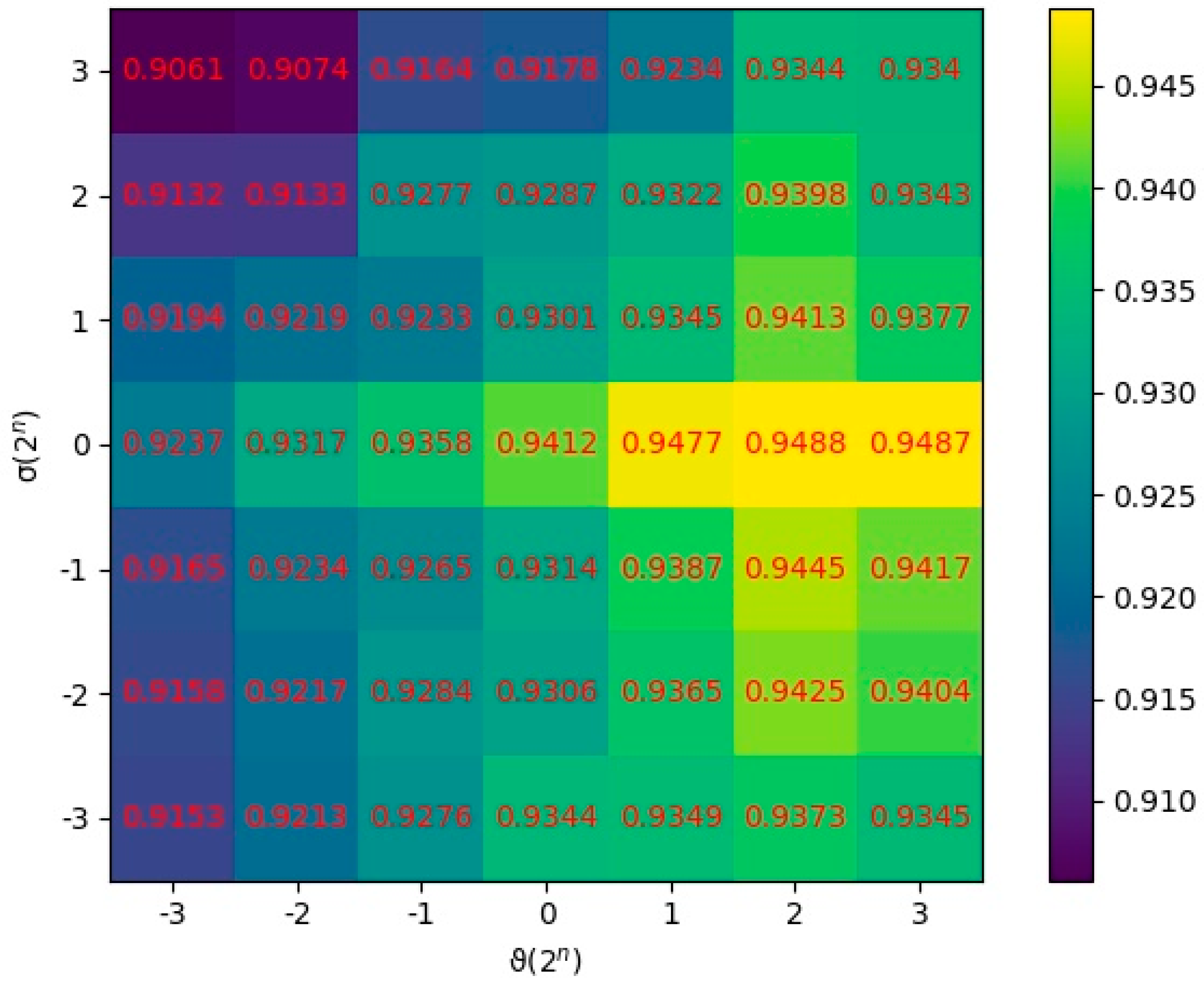
2.2. Parameter Analysis
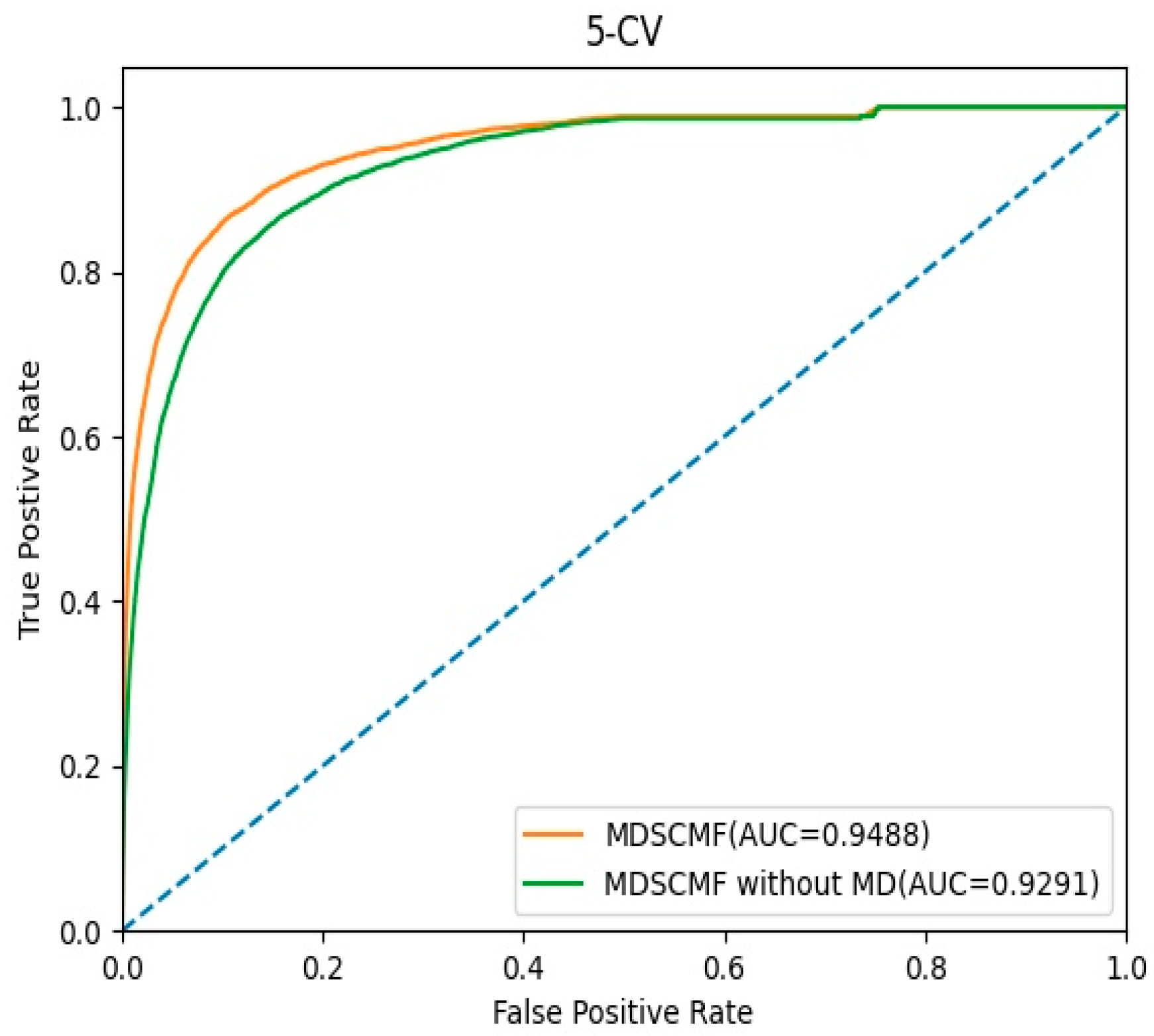
2.3. Effects of Matrix Decomposition Analysis
2.4. Case Studies
3. Materials and Methods
3.1. Human miRNA–Disease Associations
3.2. MiRNA Functional Similarity
3.3. Disease Semantic Similarity
3.4. Gaussian Interaction Profile Kernel Similarity
3.5. Integrating Similarity for miRNAs and Diseases
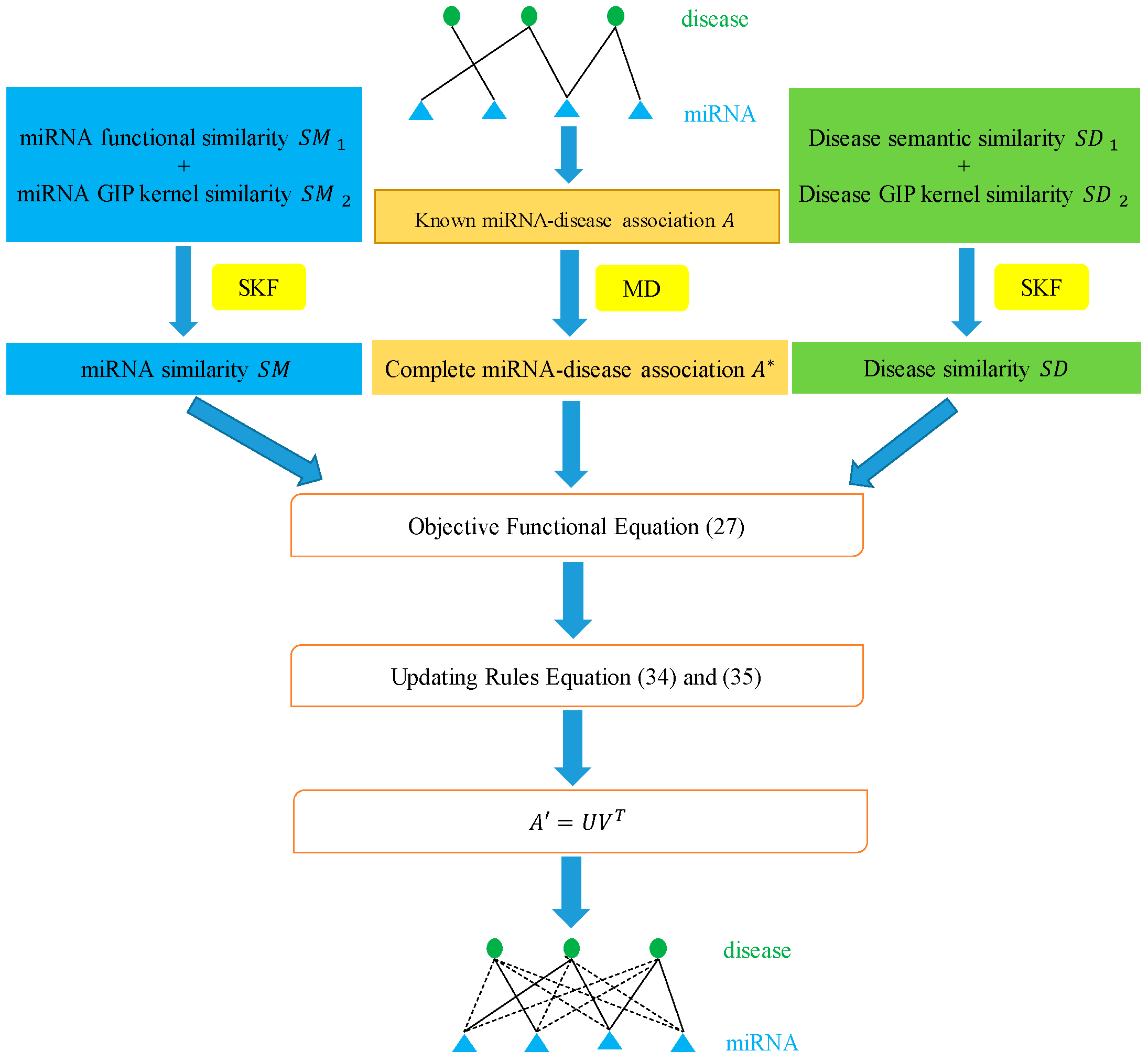
3.6. Matrix Decomposition
| Algorithm 1: Solving Equation (21) by IALM |
| Input: Given an incomplete matrix and parameters Output: and |
| Initialize:,,,,, , , while not converged do 1: Fix the other and update by 2: Fix the other and update by () 3: Fix the other and update by 4: Update the multiplier 5: Update parameter by 6: Check the convergence condition end while |
3.7. Similarity-Constrained Matrix Factorization
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vasques, L.R.; Pereira, L.V.; Izzotti, A.; Schoof, C.R.G.; Ribeiro, A.O. MicroRNAs: Modulators of cell identity, and their applications in tissue engineering. MicroRNA 2014, 3, 45–53. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Ana, K.; Sam, G.J. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, 68–73. [Google Scholar]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.M.; Byrom, M.W.; Shelton, J.; Ford, L.P. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005, 33, 1290–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, X.; Ambros, V. Developmental biology. Encountering MicroRNAs in Cell Fate Signaling. Science 2005, 310, 1288–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miska, E.A. How microRNAs control cell division, differentiation and death. Curr. Opin. Genet. Dev. 2005, 15, 563–568. [Google Scholar] [CrossRef]
- Xu, P.; Guo, M.; Hay, B.A. MicroRNAs and the regulation of cell death. Trends Genet. 2004, 20, 617–624. [Google Scholar] [CrossRef]
- Lynam-Lennon, N.; Maher, S.; Reynolds, J.V. The roles of microRNA in cancer and apoptosis. Biol. Rev. Camb. Philos. Soc. 2009, 84, 55–71. [Google Scholar] [CrossRef]
- Meola, N.; Gennarino, V.A.; Banfi, V. microRNAs and genetic diseases. PathoGenetics 2009, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Circulating microRNAs as potential new biomarkers for prostate cancer. Cancer Cell 2013, 108, 1925–1930. [Google Scholar]
- Thomson, J.M.; Parker, J.S.; Hammond, S.M. Microarray Analysis of miRNA Gene Expression. Methods Enzymol. 2007, 427, 107–122. [Google Scholar] [CrossRef]
- Han, K.; Xuan, P.; Ding, J.; Zhao, Z.J.; Hui, L.; Zhong, Y.L. Prediction of disease-related microRNAs by incorporating functional similarity and common association information. Genet. Mol. Res. 2014, 13, 2009–2019. [Google Scholar] [CrossRef]
- You, Z.; Huang, Z.; Zhu, Z.; Yan, G.; Li, Z.; Wen, Z.; Chen, X. PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 2017, 13, e1005455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Gong, Y.; Zhang, D.-H.; You, Z.; Li, Z.-W. DRMDA: Deep representations-based miRNA-disease association prediction. J. Cell. Mol. Med. 2018, 22, 472–485. [Google Scholar] [CrossRef]
- Zeng, X.; Xuan, X.; Quan, B. Integrative approaches for predicting microRNA function and prioritizing disease-related mi-croRNA using biological interaction networks. Brief Bioinform. 2016, 17, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Zhang, Q.; Min, D.; Jing, M.; Guo, Y.; Guo, W.; Cui, Q. An Analysis of Human MicroRNA and Disease Associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Hao, Y.; Wang, G.; Juan, L.; Wang, Y. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst. Biol. 2010, 4, S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, Q.; Zheng, Y.; Lv, S.; Ning, S.; Sun, J.; Huang, T.; Zheng, Q.; Ren, H.; Xu, J.; et al. Prioritizing human cancer microRNAs based on genes’ functional consistency between microRNA and cancer. Nucleic Acids Res. 2011, 39, e153. [Google Scholar] [CrossRef] [PubMed]
- Mørk, S.; Sune, P.F.; Albert, P.C.; Jan, G.; Jensen, L.J. Protein-driven inference of miRNA–disease associations. Bioinformatics 2014, 30, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yan, C.; Zhang, X.; You, Z.; Deng, L.; Liu, Y.; Zhang, Y.; Dai, Q. WBSMDA: Within and Between Score for MiRNA-Disease Association prediction. Sci. Rep. 2016, 6, 21106. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, X.; Lu, L. Large-scale prediction of microRNA-disease associations by combinatorial prioritization algorithm. Sci. Rep. 2017, 7, 43792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Cheng, J.; Yin, J. Predicting microRNA-disease associations using bipartite local models and hubness-aware regression. RNA Biol. 2018, 15, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, J.; Park, C. MLMD: Metric Learning for predicting MiRNA-Disease associations. IEEE Access. 2021, 9, 78847–78858. [Google Scholar] [CrossRef]
- Li, L.; Gao, Z.; Wang, Y.; Zhang, M.; Ni, J.; Zheng, C.; Su, Y. SCMFMDA: Predicting microRNA-disease associations based on similarity constrained matrix factorization. PLoS Comput. Biol. 2021, 17, e1009165. [Google Scholar] [CrossRef]
- Shi, H.; Xu, J.; Zhang, G.; Xu, L.; Li, C.; Wang, L.; Zhao, Z.; Jiang, W.; Guo, Z.; Li, X. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Syst. Biol. 2013, 7, 101. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, M.; Yan, G. RWRMDA: Predicting novel human microRNA-disease associations. Mol. Biosyst. 2012, 8, 2792–2798. [Google Scholar] [CrossRef]
- Köhler, S.; Bauer, S.; Horn, D.; Robinson, P.N. Walking the Interactome for Prioritization of Candidate Disease Genes. Am. J. Hum. Genet. 2008, 82, 949–958. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zeng, X.; He, Z.; Zou, Q. Inferring microRNA-disease associations by random walk on a heterogeneous network with multiple data sources. ACM Trans. Comput. Biol. Bioinform. 2017, 14, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Xiao, Q. A novel approach for predicting microRNA-disease associations by unbalanced bi-random walk on heterogeneous network. J. Biomed. Inform. 2017, 66, 194–203. [Google Scholar] [CrossRef]
- Li, J.; Rong, Z.; Chen, X.; Yan, G.; You, Z. MCMDA: Matrix completion for MiRNA-disease association prediction. Oncotarget 2017, 8, 21187–21199. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Cai, L.; Liao, B.; Zhu, W.; Yang, J. Identifying Potential miRNAs-Disease Associations with Probability Matrix Factorization. Front. Genet. 2019, 10, 1234. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Park, C.; Park, C.; Park, S. Improved Prediction of miRNA-Disease Associations Based on Matrix Completion with Network Regularization. Cells 2020, 9, 881. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Shi, K.; Lin, W. MLPMDA: Multi-layer linear projection for predicting miRNA-disease association. Knowl. Based Syst. 2021, 214, 106718. [Google Scholar] [CrossRef]
- Ding, Y.; Lei, X.; Liao, B.; Wu, F. Predicting miRNA-Disease Associations Based on Multi-View Variational Graph Auto-Encoder with Matrix Factorization. IEEE J. Biomed. Health Inform. 2021, 26, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Yue, K.; Wang, L.; Ma, Y.; Li, Q. NMCMDA: Neural multicategory MiRNA-disease association prediction. Brief Bioinform. 2021, 22, bbab074. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3. 0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wu, L.; Wang, A.; Tang, W.; Zhao, Y.; Zhao, H.; Teschendorff, A.E. dbDEMC 2.0: Updated database of differ-entially expressed miRNAs in human cancers. Nucleic Acids Res. 2017, 45, D812–D818. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Y.; Ji, C.; Zheng, C.; Ni, J.; Su, Y. GCAEMDA: Predicting miRNA-disease associations via graph convolutional. PLoS Comput. Biol. 2021, 17, e1009655. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, Y.; Gao, Z.; Ni, J.; Zheng, C. MSCHLMDA: Multi-Similarity Based Combinative Hypergraph Learning for Predicting MiRNA-Disease Association. Front. Genet. 2020, 11, 354. [Google Scholar] [CrossRef]
- Li, J.; Zhang, S.; Liu, T.; Ning, C.; Zhang, Z.; Zhou, W. Neural Inductive Matrix Completion with Graph Convolutional Networks for miRNA-disease Association Prediction. Bioinformatics 2020, 36, 2538–2546. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Q.; Gao, Z.; Ni, J.; Zheng, C. MiRNA-disease association prediction via hypergraph learning based on high-dimensionality features. BMC Med. Inform. Decis. Mak. 2021, 21, 133. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, C.; Yin, J.; You, Z. Novel Human miRNA-Disease Association Inference Based on Random Forest. Mol. Ther. -Nucleic Acids 2018, 13, 568–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Xie, D.; Wang, L.; Zhao, Q.; You, Z.; Liu, H. BNPMDA: Bipartite Network Projection for MiRNA–Disease Association prediction. Bioinformatics 2018, 34, 3178–3186. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chen, X.; Yin, J. Adaptive boosting-based computational potential miRNA-disease associations. Bioinformatics 2020, 36, 330. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Miller, K.D.; Sauer, A.G.; Jemal, A.; Siegel, R.L. Cancer statistics for African Americans, 2019. CA Cancer J. Clin. 2019, 69, S7. [Google Scholar] [CrossRef] [Green Version]
- Thackeray, E.W.; Charatcharoenwitthaya, P.; Elfaki, D.; Sinakos, E.; Lindor, K.D. Colon Neoplasms Develop Early in the Course of Inflammatory Bowel Disease and Primary Sclerosing Cholangitis. Clin. Gastroenterol. Hepatol. 2011, 9, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.W.; Chen, L.; Man, Y.-G. miRNA Biomarkers in Breast Cancer Detection and Management. J. Cancer 2011, 2, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yin, H.; Zhang, L.; Zhang, D.; Zhu, J. The construction and analysis of the aberrant lncRNA-miRNA-mRNA network in non-small cell lung cancer. J. Thorac. Dis. 2019, 11, 1772–1778. [Google Scholar] [CrossRef]
- Lipscomb, C.E. Medical Subject Headings (MeSH). Bull. Med. Libr. Assoc. 2000, 88, 265–266. [Google Scholar]
- Chen, X.; Yin, J.; Qu, J.; Huang, L.; Wang, E. MDHGI: Matrix Decomposition and Heterogeneous Graph Inference for miRNA-disease association prediction. PLoS Comput. Biol. 2018, 14, e1006418. [Google Scholar] [CrossRef]
- Xuan, P.; Han, K.; Guo, M.; Guo, Y.; Huang, Y. Prediction of microRNAs Associated with Human Diseases Based on Weighted k Most Similar Neighbors. PLoS ONE 2013, 8, e70204. [Google Scholar] [CrossRef]
- Van, L.T.; Nabuurs, S.B.; Marchiori, E. Gaussian inter-action profile kernels for predicting drug-target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar]
- Chen, X.; Huang, Y.-A.; You, Z.-H.; Yan, G.-Y.; Wang, X. A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics 2017, 33, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.W.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef]
- Yu, S.; Liang, C.; Xiao, Q.; Li, G.; Ding, P.; Luo, J. MCLPMDA: A novel method for miRNA-disease association prediction based on matrix completion and label propagation. J. Cell. Mol. Med. 2019, 23, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, J. Robust Subspace Segmentation Via Low-Rank Representation. IEEE Trans. Cybern. 2014, 44, 1432–1445. [Google Scholar] [CrossRef]
- Meng, F.; Yang, X.; Zhou, C. The Augmented Lagrange Multipliers Method for Matrix Completion from Corrupted Samplings with Application to Mixed Gaussian-Impulse Noise Removal. PLoS ONE 2014, 9, e108125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, X.R.; Chen, Y.L.; Wu, W.J.; Wang, W.; Li, X.H. Feature-derived graph regularized matrix factorization for predicting drug side effects-Science Direct. Neurocomputing 2018, 287, 154–162. [Google Scholar] [CrossRef]
- Rana, B.; Juneja, A.; Saxena, M.; Gudwani, S.; Kumaran, S.S.; Behari, M.; Agrawal, R.K. Graph-Theory-based Spectral Feature Selection for Computer Aided Diagnosis of Parkinson’s Disease Using T1-weighted MRI. Int. J. Imaging Syst. Technol. 2015, 25, 245–255. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-630 | d | hsa-mir-29b | m; d |
| hsa-mir-20a | m; d | hsa-mir-141 | m; d |
| hsa-mir-143 | m; d | hsa-mir-132 | m; d |
| hsa-mir-584 | d | hsa-mir-19b | m; d |
| hsa-mir-506 | d | hsa-mir-29a | m; d |
| hsa-mir-552 | d | hsa-mir-223 | d |
| hsa-mir-128 | unconfirmed | hsa-let-125b | d |
| hsa-mir-7i | m; d | hsa-mir-622 | d |
| hsa-mir-127 | m; d | hsa-mir-18a | d |
| hsa-mir-1290 | d | hsa-mir-143 | d |
| hsa-mir-493 | d | hsa-mir-125a | m; d |
| hsa-mir-498 | d | hsa-mir-21 | m; d |
| hsa-mir-107 | m; d | hsa-mir-137 | m; d |
| hsa-mir-191 | m; d | hsa-mir-424 | d |
| hsa-mir-32 | m; d | hsa-mir-200b | d |
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-99a | m; d | hsa-mir-663 | m |
| hsa-mir-542 | d | hsa-mir-520h | d |
| hsa-mir-96 | d | hsa-mir-519d | d |
| hsa-mir-98 | m; d | hsa-mir-186 | d |
| hsa-mir-185 | d | hsa-mir-381 | d |
| hsa-mir-130a | d | hsa-mir-32 | d |
| hsa-mir-708 | d | hsa-mir-590 | unconfirmed |
| hsa-mir-150 | d | hsa-mir-330 | d |
| hsa-mir-192 | d | hsa-mir-433 | d |
| hsa-mir-196b | d | hsa-mir-942 | d |
| hsa-mir-888 | d | hsa-mir-661 | m; d |
| hsa-mir-9 | m; d | hsa-mir-337 | d |
| hsa-mir-130b | d | hsa-mir-494 | d |
| hsa-mir-592 | d | hsa-mir-212 | d |
| hsa-mir-99b | d | hsa-mir-618 | d |
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-96 | d | hsa-mir-937 | unconfirmed |
| hsa-mir-145 | m; d | hsa-mir-30e | m |
| hsa-mir-99a | m; d | hsa-mir-151 | d |
| hsa-mir-9 | m; d | hsa-mir-614 | d |
| hsa-mir-185 | d | hsa-mir-1323 | d |
| hsa-mir-130a | d | hsa-mir-32 | d |
| hsa-mir-7 | m; d | hsa-mir-1298 | d |
| hsa-mir-150 | m; d | hsa-mir-330 | d |
| hsa-mir-192 | m; d | hsa-mir-433 | d |
| hsa-mir-769 | unconfirmed | hsa-mir-522 | d |
| hsa-mir-939 | d | hsa-mir-449a | d |
| hsa-mir-98 | m; d | hsa-mir-143 | m; d |
| hsa-mir-130b | m; d | hsa-mir-564 | d |
| hsa-mir-638 | d | hsa-mir-212 | m; d |
| hsa-mir-99b | d | hsa-mir-615 | unconfirmed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, J.; Li, L.; Wang, Y.; Ji, C.; Zheng, C. MDSCMF: Matrix Decomposition and Similarity-Constrained Matrix Factorization for miRNA–Disease Association Prediction. Genes 2022, 13, 1021. https://doi.org/10.3390/genes13061021
Ni J, Li L, Wang Y, Ji C, Zheng C. MDSCMF: Matrix Decomposition and Similarity-Constrained Matrix Factorization for miRNA–Disease Association Prediction. Genes. 2022; 13(6):1021. https://doi.org/10.3390/genes13061021
Chicago/Turabian StyleNi, Jiancheng, Lei Li, Yutian Wang, Cunmei Ji, and Chunhou Zheng. 2022. "MDSCMF: Matrix Decomposition and Similarity-Constrained Matrix Factorization for miRNA–Disease Association Prediction" Genes 13, no. 6: 1021. https://doi.org/10.3390/genes13061021