
Exploring Relevant mRNAs and miRNAs in Injured Urethral Tissues of Rats with High-Throughput Sequencing

 ,
, 
 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Establishment of Urethral Injury Animal Model
2.3. Tissue Collection
2.4. RNA Isolation, Library Preparation and Sequencing
2.5. Generation and Analysis of the RNA-seq Profiles in Rat Urethra
2.6. Gene Set Enrichment Analysis
2.7. Small RNA Sequencing Profiles
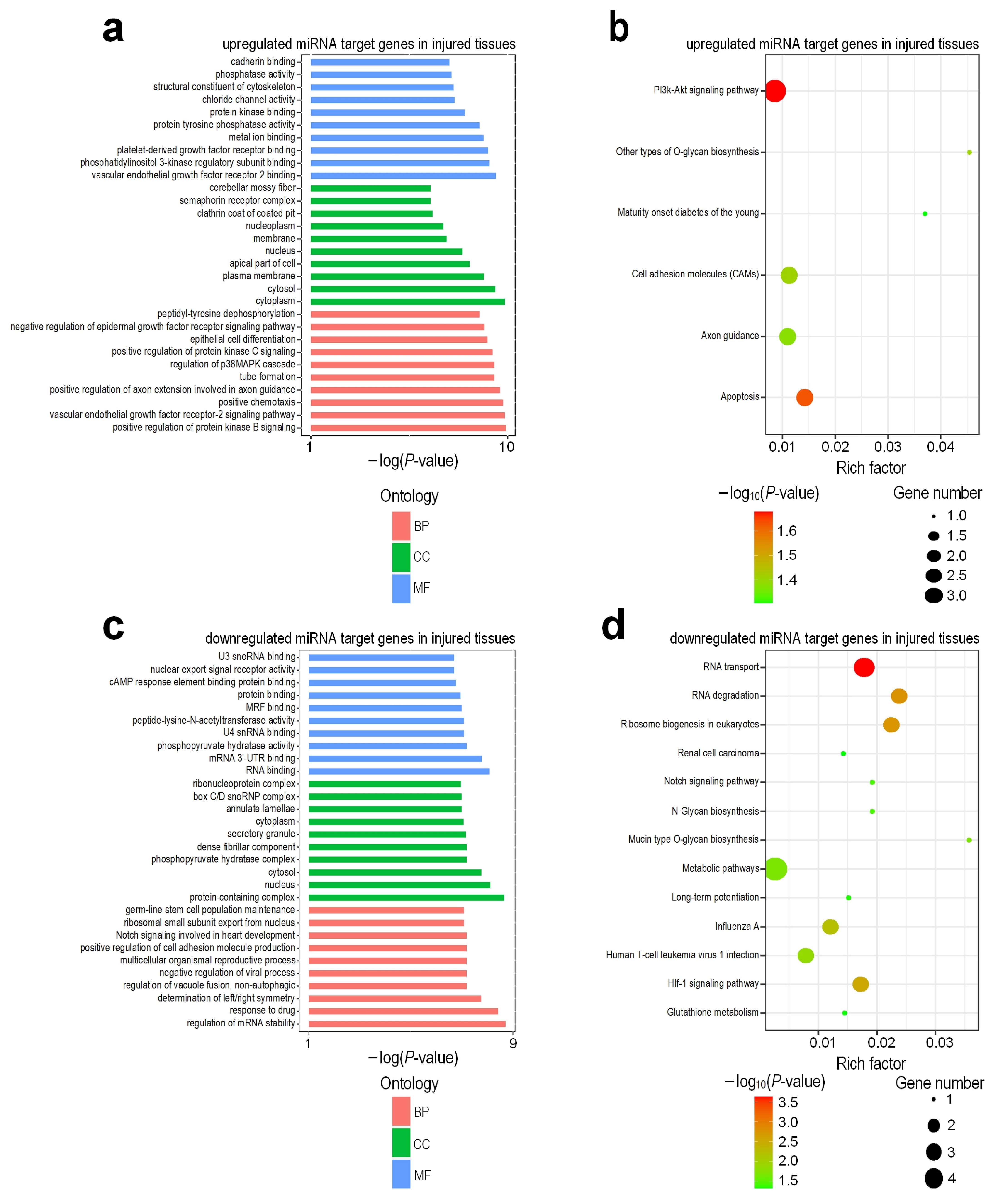
2.8. Identification of miRNA Targets and GO and KEGG Enrichment Analysis
3. Results
3.1. Quality Examination of RNA-seq Data
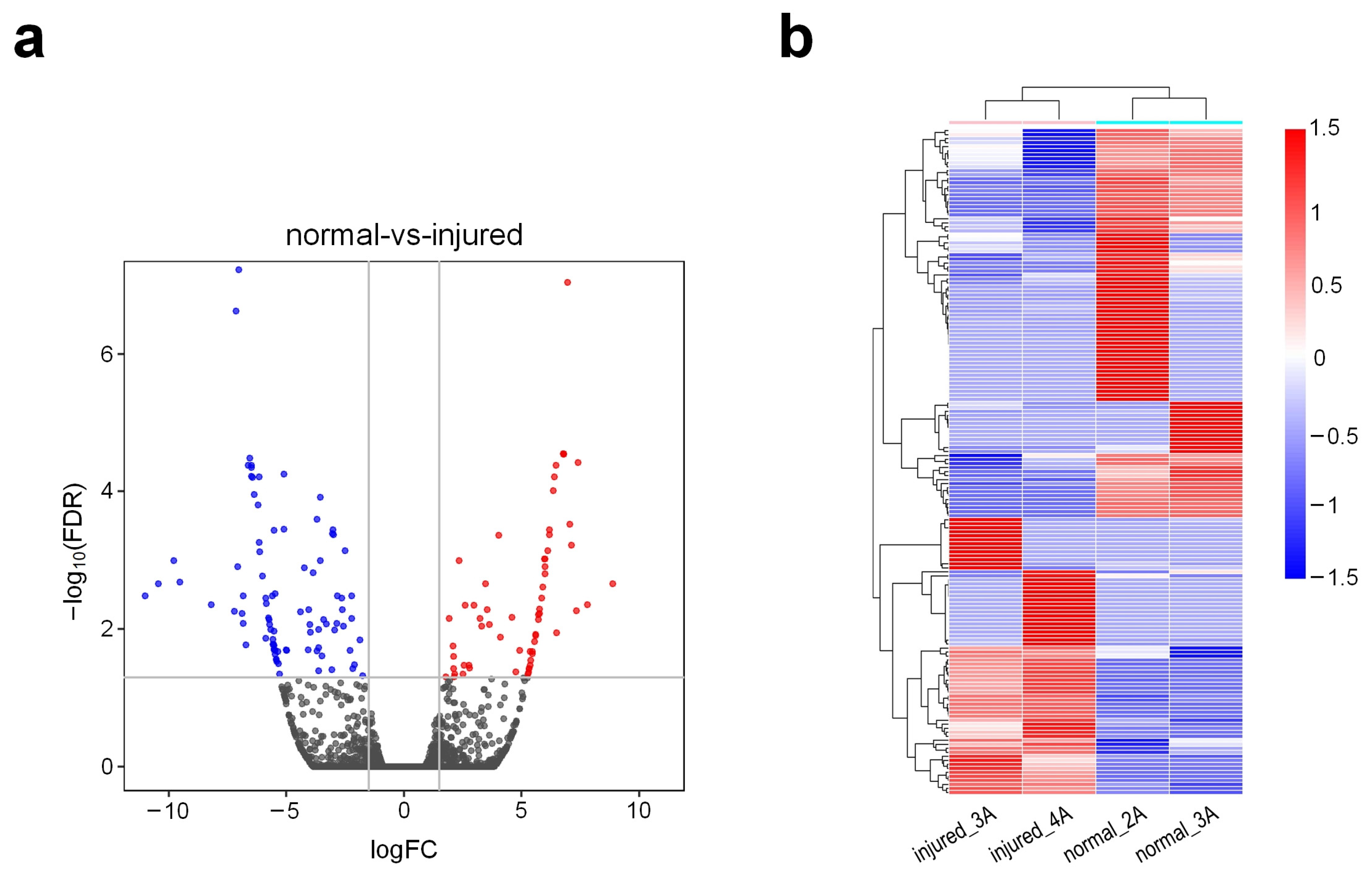
3.2. Gene Expression Patterns of Injured Rat Urethra
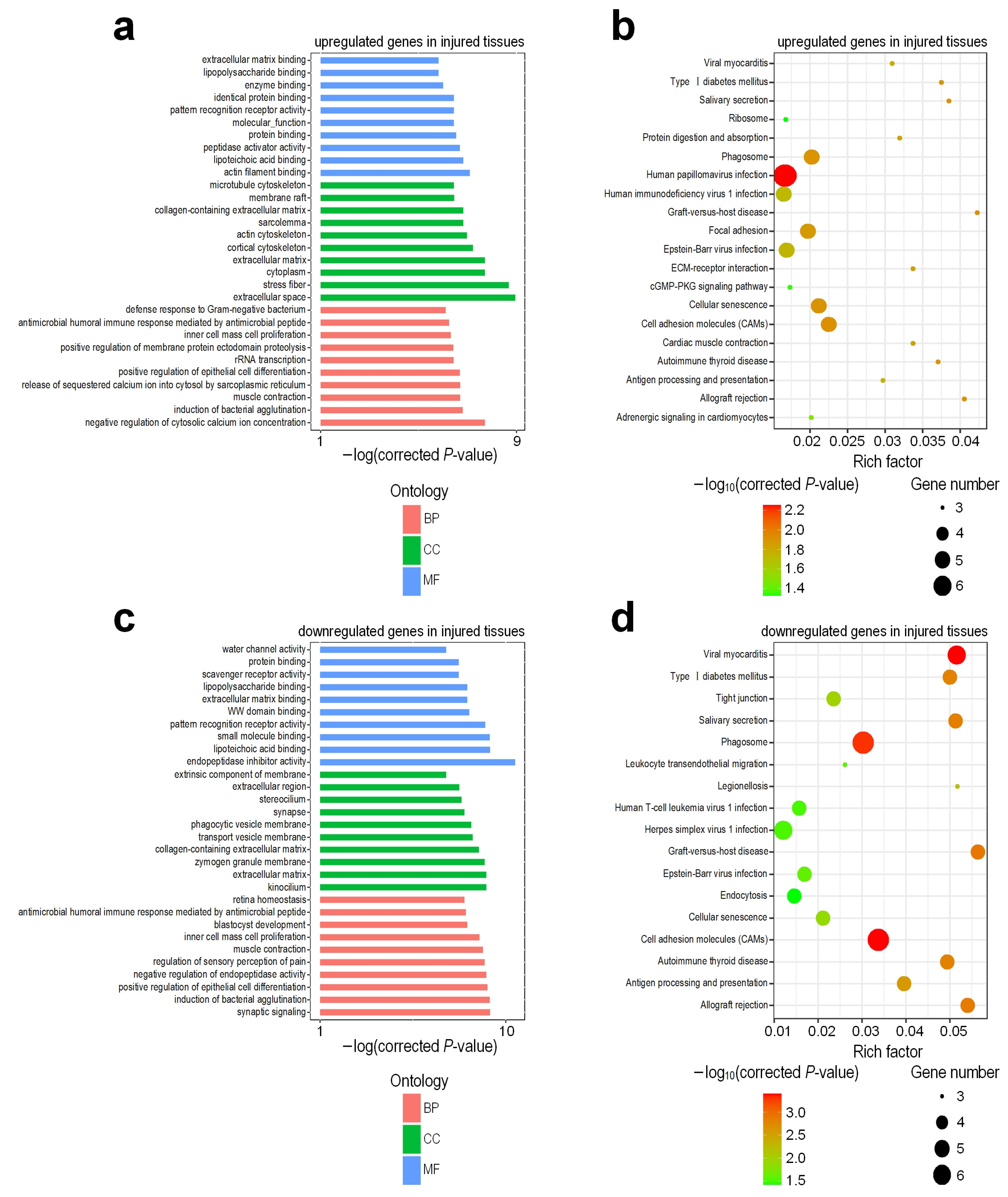
3.3. Functional Enrichment Analysis of Differentially Expressed Genes
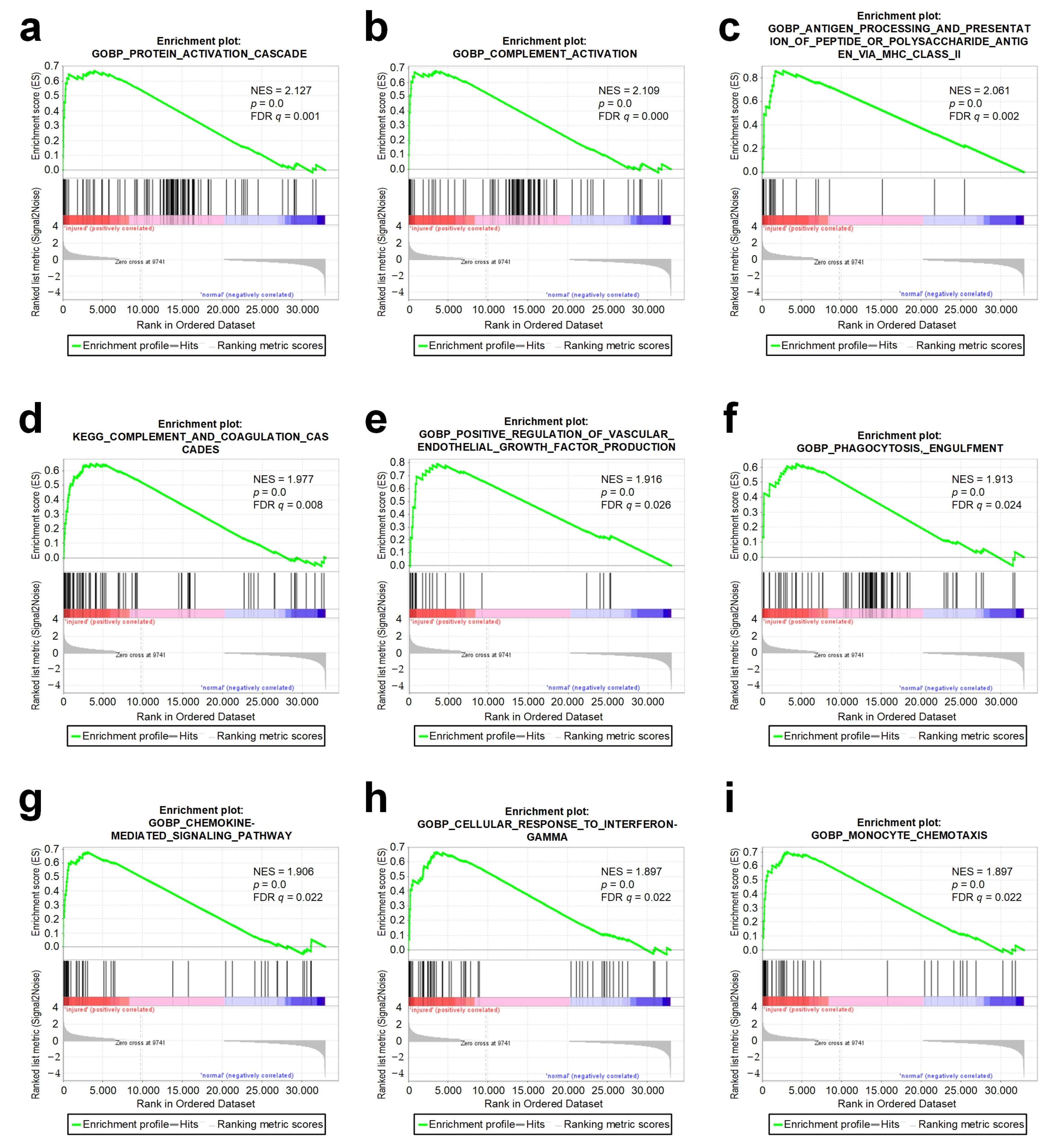
3.4. The GSEA Analysis
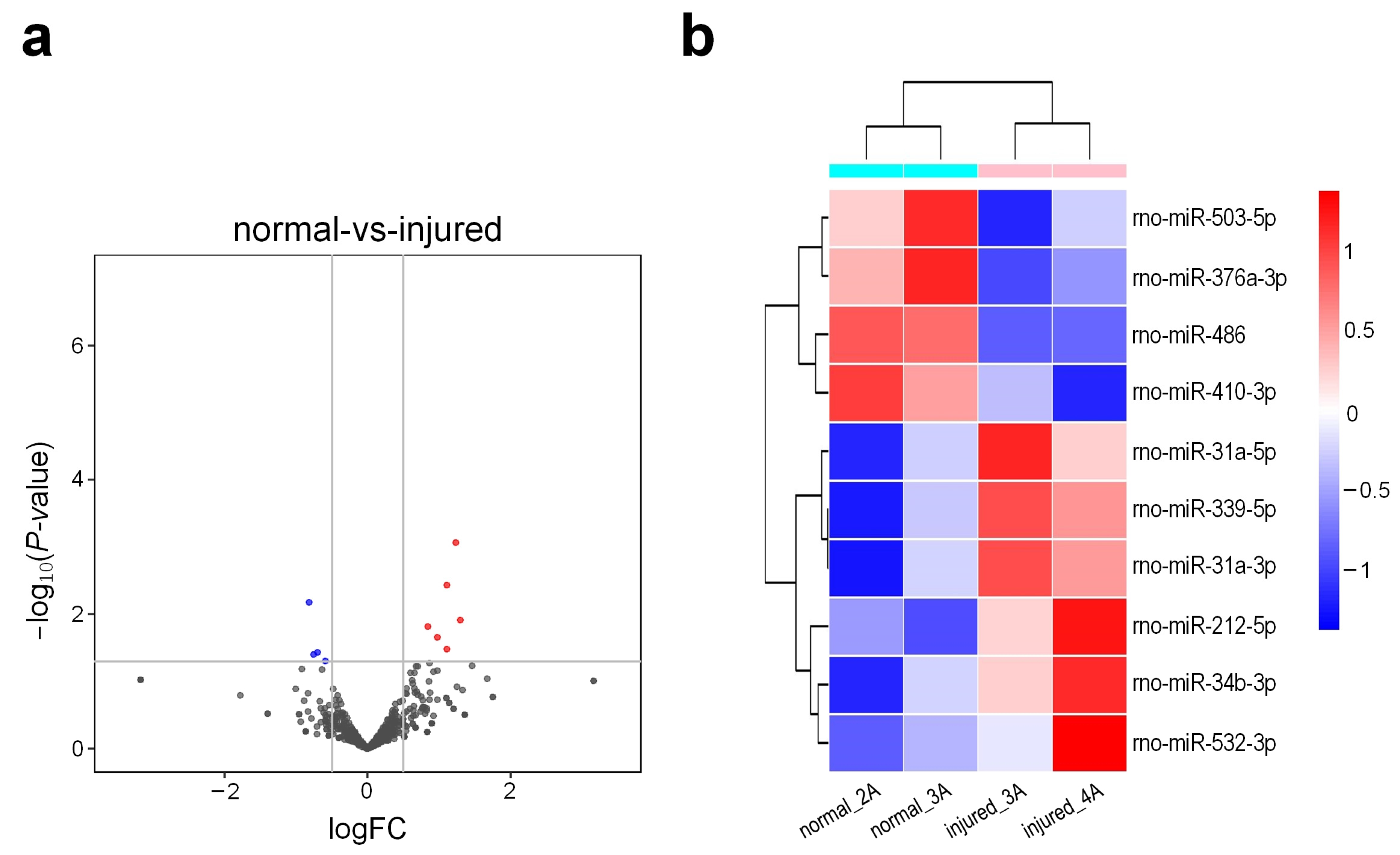
3.5. Small RNA Sequencing Profiles of Different Tissues of Rat Urethra
3.6. Conserved miRNAs and Their Expression Patterns in Different Tissues
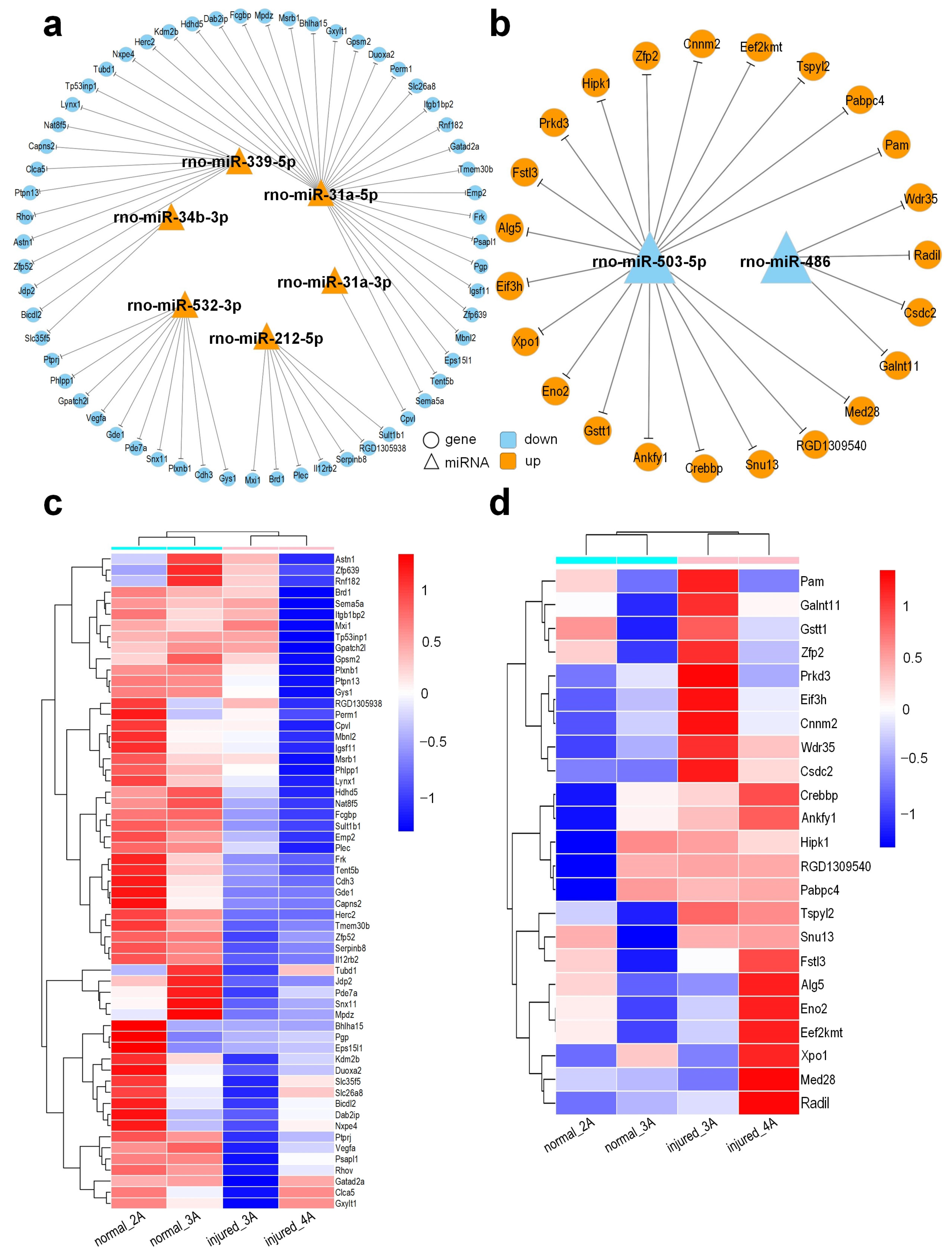
3.7. Target Gene Prediction and Functional Regulatory Network of DEMs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kashefi, C.; Messer, K.; Barden, R.; Sexton, C.; Parsons, J.K. Incidence and Prevention of Iatrogenic Urethral Injuries. J. Urol. 2008, 179, 2254–2258. [Google Scholar] [CrossRef] [PubMed]
- Hadjizacharia, P.; Inaba, K.; Teixeira, P.G.; Kokorowski, P.; Demetriades, D.; Best, C. Evaluation of immediate endoscopic realignment as a treatment modality for traumatic urethral injuries. J. Trauma Acute Care Surg. 2008, 64, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Rieder, J.; Brusky, J.; Tran, V.; Stern, K.; Aboseif, S. Review of intentionally self-inflicted, accidental and iatrogetic foreign objects in the genitourinary tract. Urol. Int. 2010, 84, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, G.G.; Bullock, T.L.; Anderson, R.E.; Blalock, R.E.; Brandes, S.B. Minimally invasive methods for bulbar urethral strictures: A survey of members of the American Urological Association. Urology 2011, 78, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Akyuz, M.; Sertkaya, Z.; Koca, O.; Caliskan, S.; Kutluhan, M.A.; Karaman, M.I. Adult urethral stricture: Practice of Turkish urologists. Int. Braz. J. Urol. 2016, 42, 339–345. [Google Scholar] [CrossRef]
- Greenwell, T.J.; Castle, C.; Andrich, D.E.; MacDonald, J.T.; Nicol, D.L.; Mundy, A.R. Repeat urethrotomy and dilation for the treatment of urethral stricture are neither clinically effective nor cost-effective. J. Urol. 2004, 172, 275–277. [Google Scholar] [CrossRef]
- Mundy, A.R.; Andrich, D.E. Urethral strictures. BJU Int. 2011, 107, 6–26. [Google Scholar] [CrossRef]
- Santucci, R.A.; Joyce, G.F.; Wise, M. Male urethral stricture disease. J. Urol. 2007, 177, 1667–1674. [Google Scholar] [CrossRef]
- Chen, C.; Zeng, M.; Xue, R.; Wang, G.; Gao, Z.; Yuan, W.; Tang, Z. Causes and management for male urethral stricture. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2018, 43, 520–527. [Google Scholar] [CrossRef]
- Gonzalez, A.C.; Costa, T.F.; Andrade, Z.A.; Medrado, A.R. Wound healing—A literature review. An. Bras. Dermatol. 2016, 91, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chen, J.; Kirsner, R. Pathophysiology of acute wound healing. Clin. Dermatol. 2007, 25, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Conner, E.M.; Grisham, M.B. Inflammation, free radicals, and antioxidants. Nutrition 1996, 12, 274–277. [Google Scholar] [CrossRef]
- Gilman, K.E.; Limesand, K.H. The complex role of prostaglandin E(2)-EP receptor signaling in wound healing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021, 320, R287–R296. [Google Scholar] [CrossRef] [PubMed]
- Medrado, A.R.; Pugliese, L.S.; Reis, S.R.; Andrade, Z.A. Influence of low level laser therapy on wound healing and its biological action upon myofibroblasts. Lasers Surg. Med. 2003, 32, 239–244. [Google Scholar] [CrossRef]
- Baskin, L.S.; Constantinescu, S.C.; Howard, P.S.; McAninch, J.W.; Ewalt, D.H.; Duckett, J.W.; Snyder, H.M.; Macarak, E.J. Biochemical characterization and quantitation of the collagenous components of urethral stricture tissue. J. Urol. 1993, 150, 642–647. [Google Scholar] [CrossRef]
- Hofer, M.D.; Cheng, E.Y.; Bury, M.I.; Park, E.; Xu, W.; Hong, S.J.; Kaplan, W.E.; Sharma, A.K. Analysis of primary urethral wound healing in the rat. Urology 2014, 84, 246.e1–246.e7. [Google Scholar] [CrossRef]
- Xie, H.; Feng, C.; Fu, Q.; Sa, Y.L.; Xu, Y.M. Crosstalk between TGF-β1 and CXCR3 signaling during urethral fibrosis. Mol. Cell Biochem. 2014, 394, 283–290. [Google Scholar] [CrossRef]
- Mardis, E.R. Next-generation DNA sequencing methods. Annu. Rev. Genom. Hum. Genet. 2008, 9, 387–402. [Google Scholar] [CrossRef] [Green Version]
- Schulte, L.N.; Bertrams, W.; Stielow, C.; Schmeck, B. ncRNAs in Inflammatory and Infectious Diseases. Methods Mol. Biol. 2019, 1912, 3–32. [Google Scholar] [CrossRef]
- Ying, S.Y.; Chang, D.C.; Lin, S.L. The MicroRNA. Methods Mol. Biol. 2018, 1733, 1–25. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Zhang, C.; Zhan, Q.; An, F.; Zhu, W.; Jiang, H.; Ma, C. Profiling circRNA and miRNA of radiation-induced esophageal injury in a rat model. Sci. Rep. 2018, 8, 14605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, P.C.; Chen, C.C.; Chen, Y.C.; Chang, Y.S.; Chu, P.H. MicroRNAs in acute kidney injury. Hum. Genom. 2016, 10, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, C.; Liu, J.; Dong, S. miRNA-214 Protects Sepsis-Induced Myocardial Injury. Shock 2018, 50, 112–118. [Google Scholar] [CrossRef]
- Liu, N.K.; Wang, X.F.; Lu, Q.B.; Xu, X.M. Altered microRNA expression following traumatic spinal cord injury. Exp. Neurol. 2009, 219, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y. Computational Non-Coding RNA Biology; Academic Press: Cambridge, MA, USA, 2018; pp. 35–82. [Google Scholar]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2018, 47, D155–D162. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, W. Animal microrna target prediction using diverse sequence-specific determinants. J. Bioinform. Comput. Biol. 2010, 8, 763–788. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Russo, M.V.; McGavern, D.B. Inflammatory neuroprotection following traumatic brain injury. Science 2016, 353, 783–785. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latz, E. The inflammasomes: Mechanisms of activation and function. Curr. Opin. Immunol. 2010, 22, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Li, J.; Lin, M.; Zhang, S.; Luo, J.; Tang, Y. An RNA-seq-Based Expression Profiling of Radiation-Induced Esophageal Injury in a Rat Model. Dose Response 2019, 17, 1559325819843373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holers, V.M. Complement and Its Receptors: New Insights into Human Disease. Annu. Rev. Immunol. 2014, 32, 433–459. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.C.; Isenman, D.E. Regulation of humoral immunity by complement. Immunity 2012, 37, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef]
- Sinno, H.; Malhotra, M.; Lutfy, J.; Jardin, B.; Winocour, S.; Brimo, F.; Beckman, L.; Watters, K.; Philip, A.; Williams, B.; et al. Accelerated wound healing with topical application of complement C5. Plast. Reconstr. Surg. 2012, 130, 523–529. [Google Scholar] [CrossRef]
- Wang, R.; Xiao, H.; Guo, R.; Li, Y.; Shen, B. The role of C5a in acute lung injury induced by highly pathogenic viral infections. Emerg. Microbes Infect. 2015, 4, e28. [Google Scholar] [CrossRef]
- Peuget, S.; Bonacci, T.; Soubeyran, P.; Iovanna, J.; Dusetti, N.J. Oxidative stress-induced p53 activity is enhanced by a redox-sensitive TP53INP1 SUMOylation. Cell Death Differ. 2014, 21, 1107–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Huang, H.; Xie, Q.; Wang, Z.; Fan, Y.; Kong, B.; Huang, D.; Xiao, Y. MiR-155 Knockout in Fibroblasts Improves Cardiac Remodeling by Targeting Tumor Protein p53-Inducible Nuclear Protein 1. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Yin, X.; Lai, X.; Liu, C.; Nie, W.; Li, D.; Xie, Z.; Li, Z.; Meng, F. Upregulation of DAB2IP Inhibits Ras Activity and Tumorigenesis in Human Pancreatic Cancer Cells. Technol. Cancer Res. Treat. 2020, 19, 1533033819895494. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, Q.; Liang, Y.; Pan, W.; Bei, Y.; Zhang, Y.; Wang, J.; Jiao, Z. miR-486 inhibits PM2.5-induced apoptosis and oxidative stress in human lung alveolar epithelial A549 cells. Ann. Transl. Med. 2018, 6, 209. [Google Scholar] [CrossRef]
- Xiao, Y. MiR-486-5p inhibits the hyperproliferation and production of collagen in hypertrophic scar fibroblasts via IGF1/PI3K/AKT pathway. J. Dermatol. Treat. 2020, 32, 973–982. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, H.; Guo, S.; Li, S.; Shen, J.; He, J.; Zheng, Y.; Gao, Z. Exploring Relevant mRNAs and miRNAs in Injured Urethral Tissues of Rats with High-Throughput Sequencing. Genes 2022, 13, 824. https://doi.org/10.3390/genes13050824
Lin H, Guo S, Li S, Shen J, He J, Zheng Y, Gao Z. Exploring Relevant mRNAs and miRNAs in Injured Urethral Tissues of Rats with High-Throughput Sequencing. Genes. 2022; 13(5):824. https://doi.org/10.3390/genes13050824
Chicago/Turabian StyleLin, Han, Shiyong Guo, Song Li, Jihong Shen, Jianfeng He, Yun Zheng, and Zhenhua Gao. 2022. "Exploring Relevant mRNAs and miRNAs in Injured Urethral Tissues of Rats with High-Throughput Sequencing" Genes 13, no. 5: 824. https://doi.org/10.3390/genes13050824