
A Series of 14 Polish Patients with Thrombotic Events and PC Deficiency-Novel c.401-1G>A PROC Gene Splice Site Mutation in a Patient with Aneurysms


 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Blood Samples
2.3. Laboratory Tests
2.4. Genetic Analysis
2.5. Statistical Analysis
3. Results
3.1. PROC Mutations
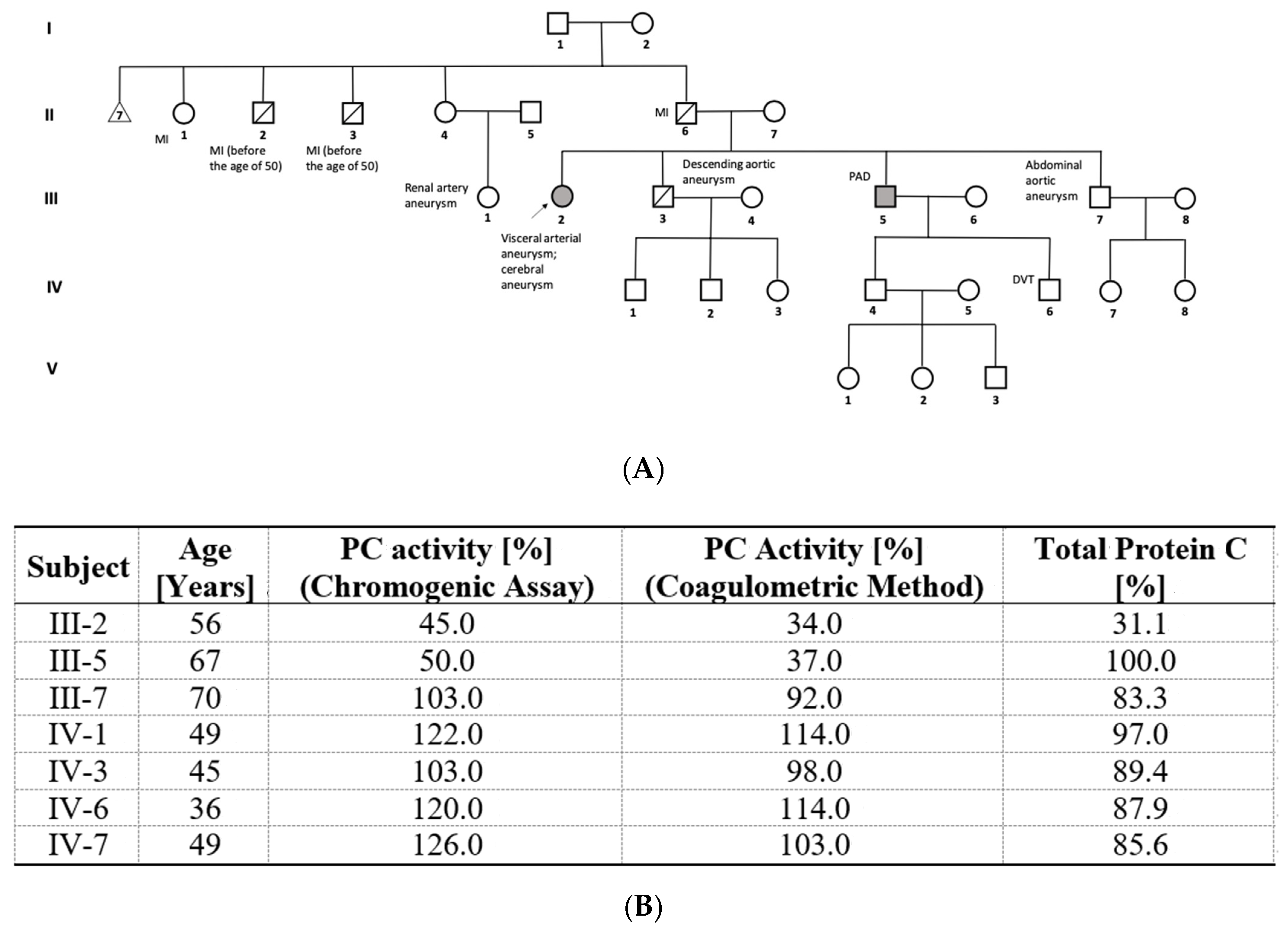
3.2. Novel PROC Gene Splice Site Mutation and Aneurysms
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dinarvand, P.; Moser, K.A. Protein C Deficiency. Arch. Pathol. Lab. Med. 2019, 143, 1281–1285. [Google Scholar] [CrossRef] [Green Version]
- Brummel-Ziedins, K.E.; Orfeo, T.; Callas, P.W.; Gissel, M.; Mann, K.G.; Bovill, E.G. The prothrombotic phenotypes in familial protein C deficiency are differentiated by computational modeling of thrombin generation. PLoS ONE 2012, 7, e44378. [Google Scholar] [CrossRef]
- Caspers, M.; Pavlova, A.; Driesen, J.; Harbrecht, U.; Klamroth, R.; Kadar, J.; Fischer, R.; Kemkes-Matthes, B.; Oldenburg, J. Deficiencies of antithrombin, protein C and protein S—Practical experience in genetic analysis of a large patient cohort. Thromb. Haemost. 2012, 108, 247–257. [Google Scholar] [CrossRef]
- España, F.; Vayá, A.; Mira, Y.; Medina, P.; Estellés, A.; Villa, P.; Falcó, C.; Aznar, J. Low level of circulating activated protein C is a risk factor for venous thromboembolism. Thromb. Haemost. 2001, 86, 1368–1373. [Google Scholar]
- Bereczky, Z.; Kovács, K.B.; Muszbek, L. Protein C and protein S deficiencies: Similarities and differences between two brothers playing in the same game. Clin. Chem. Lab. Med. 2010, 1, S53–S66. [Google Scholar] [CrossRef] [Green Version]
- Mahmoodi, B.K.; Brouwer, J.L.; Veeger, N.J.; Van der Meer, J. Hereditary deficiency of protein C or protein S confers increased risk of arterial thromboembolic events at a young age: Results from a large family cohort study. Circulation 2008, 118, 1659–1667. [Google Scholar] [CrossRef] [Green Version]
- Wypasek, E.; Pankiw-Bembenek, O.; Potaczek, D.P.; Alhenc-Gelas, M.; Trebacz, J.; Undas, A. A missense mutation G109R in the PROC gene associated with type I protein C deficiency in a young Polish man with acute myocardial infarction. Int. J. Cardiol. 2013, 167, 146–148. [Google Scholar] [CrossRef]
- Zorio, E.; Navarro, S.; Medina, P.; Estellés, A.; Osa, A.; Rueda, J.; Cubillo, P.; Aznar, J.; España, F. Circulating activated protein C is reduced in young survivors of myocardial infarction and inversely correlates with the severity of coronary lesions. J. Thromb. Haemost. 2006, 4, 1530–1536. [Google Scholar] [CrossRef]
- Wypasek, E.; Corral, J.; Alhenc-Gelas, M.; Sydor, W.; Iwaniec, T.; Celińska-Lowenhoff, M.; Potaczek, D.P.; Blecharczyk, A.; Zawilska, K.; Musiał, J.; et al. Genetic characterization of antithrombin, protein C, and protein S deficiencies in Polish patients. Pol. Arch. Intern. Med. 2017, 127, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Wypasek, E.; Potaczek, D.P.; Alhenc-Gelas, M.; Undas, A. Novel missense mutation C106R in the PROC gene associated with type I protein C deficiency in a young Polish man with high-risk pulmonary embolism. Pol. Arch. Intern. Med. 2014, 124, 75–76. [Google Scholar] [CrossRef] [Green Version]
- Schulman, S.; Angerås, U.; Bergqvist, D.; Eriksson, B.; Lassen, M.R.; Fisher, W. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in surgical patients. J. Thromb. Haemost. 2010, 8, 202–204. [Google Scholar] [CrossRef]
- Martos, L.; Fernández-Pardo, Á.; López-Fernández, M.F.; Ibáñez, F.; Herrero, S.; Tàssies, D.; González-Porras, J.R.; Solmoirago, M.J.; Costa, M.J.; Reverter, J.C.; et al. Identification of 58 Mutations (26 Novel) in 94 of 109 Symptomatic Spanish Probands with Protein C Deficiency. Thromb. Haemost. 2019, 119, 1409–1418. [Google Scholar] [CrossRef]
- Speker, M.; Balogh, G.; Pfliegler, G. Genetic heterogeneity of protein C deficiency in Hungary; genotype-phenotype correlations. ISTH 2017, 22, 2017. [Google Scholar]
- Campens, L.; Callewaert, B.; Muiño Mosquera, L.; Renard, M.; Symoens, S.; De Paepe, A.; Coucke, P.; De Backer, J. Gene panel sequencing in heritable thoracic aortic disorders and related entities—Results of comprehensive testing in a cohort of 264 patients. Orphanet J. Rare Dis. 2015, 10, 9. [Google Scholar] [CrossRef]
- Ito, K.; Patel, P.N.; Gorham, J.M.; McDonough, B.; DePalma, S.R.; Adler, E.E.; Lam, L.; MacRae, C.A.; Mohiuddin, S.M.; Fatkin, D.; et al. Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proc. Natl. Acad. Sci. USA 2017, 114, 7689–7694. [Google Scholar] [CrossRef] [Green Version]
- Andreou, A.; Papapetrou, C.; Papadimitriou, K.; Avgoustidis, D.; Yapijakis, C. Cerebrovascular Aneurysms May Be Associated with Thrombophilia-predisposing Mutations in Patients with Familial Risk. In Vivo 2015, 29, 395–398. [Google Scholar]
- Ozge, C.; Calikoğlu, M.; Yildiz, A.; Türsen, U.; Tamer, L. Bilateral pulmonary artery aneurysms with protein C and protein S deficiency in a patient with Behçet’s disease. Scand. J. Rheumatol. 2004, 33, 52–54. [Google Scholar] [CrossRef]
- Navarro, S.; Ricart, J.M.; Medina, P.; Vayá, A.; Villa, P.; Todolí, J.; Estellés, A.; Micó, M.L.; Aznar, J.; España, F. Activated protein C levels in Behçet’s disease and risk of venous thrombosis. Br. J. Haematol. 2004, 126, 550–556. [Google Scholar] [CrossRef]
- Navarro, S.; Bonet, E.; Medina, P.; Martos, L.; Ricart, J.M.; Vayá, A.; Todolí, J.; Fontcuberta, J.; Estellés, A.; España, F. Haplotypes of the endothelial protein C receptor gene and Behçet’s disease. Thromb. Res. 2012, 129, 459–464. [Google Scholar] [CrossRef]
- Canpolat, U.; Yayla, Ç.; Çetin, E.H.; Çay, S.; Aras, D. Unpredictable coupling: Thrombophilia in a patient with Marfan syndrome. Blood Coagul. Fibrinolysis 2015, 26, 713–714. [Google Scholar] [CrossRef]
- De la Morena-Barrio, M.E.; Gindele, R.; Bravo-Pérez, C.; Ilonczai, P.; Zuazu, I.; Speker, M.; Oláh, Z.; Rodríguez-Sevilla, J.J.; Entrena, L.; Infante, M.S.; et al. High penetrance of inferior vena cava system atresia in severe thrombophilia caused by homozygous antithrombin Budapest 3 variant: Description of a new syndrome. Am. J. Hematol. 2021, 96, 1363–1373. [Google Scholar] [CrossRef]
- Dahlbäck, B.; Villoutreix, B.O. Molecular recognition in the protein C anticoagulant pathway. J. Thromb. Haemost. 2003, 1, 1525–1534. [Google Scholar] [CrossRef] [Green Version]
- Undas, A.; Goralczyk, T. Non-vitamin K antagonist oral anticoagulants in patients with severe inherited thrombophilia: A series of 33 patients. Blood Coagul. Fibrinolysis 2017, 28, 438–442. [Google Scholar] [CrossRef]
- Alhenc-Gelas, M.; Plu-Bureau, G.; Mauge, L.; Gandrille, S.; Présot, I.; GFHT Study Group on Genetic Thrombophilia. Genotype-Phenotype Relationships in a Large French Cohort of Subjects with Inherited Protein C Deficiency. Thromb. Haemost. 2020, 120, 1270–1281. [Google Scholar] [CrossRef]


{kind=link}
| Patient ID | Sex/Age | PC Activity % (Chromogenic Assay) | PC Activity % (Clot-Based Assay) | Total PC % | Type of PC Deficiency | Type of Mutation in PROC Gene | Exon Number | New/Reported | Clinical Manifestation | Age of First Thromboembolic Event | Number of VTE Events | Unprovoked/Provoked | Family History of VTE | Duration (Months) | Thrombo Embolic Events | Antithrombotic Treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F/56 | 45 | 34 | N/D | I | c.401-1G>A | Intron 5 | New | Hepatic artery aneurysm | 51 | N/A | N/A | 1 | 54 | 0 | ASA (75 mg/day) |
| 2 | M/54 | 65 | 56 | 118 | II | c.759C>A, p.His253Gln | Exon 8 | Reported | DVT | 50 | 1 | 0/long journey | 0 | 57 | 0 | Dabigatran (150 mg bid) |
| 3 | F/44 | 56 | N/D | N/D | II | c.759C>A, p.His253Gln | Exon 8 | Reported | DVT | 37 | 1 | 1/0 | 0 | 84 | 0 | Sulodexid (2 × 250 SLU) |
| 4 | F/35 | 68 | 64 | 139 | IIa | c.759C>A, p.His253Gln | Exon 8 | Reported | DVT-cesarean section complicated with hemorrhage | 33 | 1 | 0/pregnancy | 1 | 7 | 0 | Dabigatran (150 mg bid) |
| 5 | M/52 | 68 | 59 | 54 | II | c.759C>A, p.His253Gln | Exon 8 | Reported | DVT | 47 | 2 | 1/0 | 0 | 18 | 0 | Rivaroxaban (15 mg/day) |
| 6 | M/63 | 58 | 45 | 113 | II | c.759C>A; p.His253Gln | Exon 8 | Reported | DVT+PE | 62 | 1 | 0/long journey | 1 | 5 | 0 | Rivaroxaban (20 mg/day) |
| 7 | F/64 | 67 | 57 | 46 | I | c.400+2T>C | Intron 5 | Reported | Ischemic stroke | 59 | 0 | 1/0 | 1 | 54 | 0 | Clopidogrel (75 mg/day) |
| 8 | F/39 | 72 | 50 | N/D | N/A | c.1042C>T, p.Arg348 * | Exon 9 | Reported | Asymptomatic | N/A | N/A | N/A | 1 | 36 | 0 | none |
| 9 | M/22 | 47 | 39 | 53 | I | c.316T>C, p.Cys106Arg | Exon 5 | Reported | DVT | 20 | 1 | 1/0 | 0 | 13 | 0 | Dabigatran (150 mg bid) |
| 10 | F/48 | 46 | 34 | 35 | I | c.316T>C, p.Cys106Arg | Exon 5 | Reported | DVT+PE | 45 | 1 | 0/surgery | 0 | 31 | 0 | Apixaban (2.5 mg bid) |
| 11 | F/32 | 59 | 37 | 103 | II | c.595C>T, p.Arg199 * | Exon 7 | Reported | DVT+PE | 30 | 1 | 0/oral contraceptives | 0 | 26 | 0 | Rivaroxaban (20 mg/day) |
| 12 | F/26 | 52; 56 | 50 | 54 | I | c.962C>T, p.Pro321Leu | Exon 9 | Reported | VTE | 25 | 1 | 0/oral contraceptives | 1 | 12 | 0 | Apixaban (5 mg bid) |
| 13 | F/43 | 68 | 52 | 66.9 | I | c.1174G>A p.Gly392Arg | Exon 9 | Reported | PE | 42 | 1 | 0/oral contraceptives | 1 | 6 | 0 | Apixaban (2.5 mg bid) |
| 14 | M/37 | 56 | 46 | 69.8 | I | c.632G>A p.Arg211Gln | Exon 7 | Reported | SVT | 36 | 1 | 1/0 | 0 | 6 | 0 | Rivaroxaban (20 mg/day) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weronska, A.; Potaczek, D.P.; Oto, J.; Medina, P.; Undas, A.; Wypasek, E. A Series of 14 Polish Patients with Thrombotic Events and PC Deficiency-Novel c.401-1G>A PROC Gene Splice Site Mutation in a Patient with Aneurysms. Genes 2022, 13, 733. https://doi.org/10.3390/genes13050733
Weronska A, Potaczek DP, Oto J, Medina P, Undas A, Wypasek E. A Series of 14 Polish Patients with Thrombotic Events and PC Deficiency-Novel c.401-1G>A PROC Gene Splice Site Mutation in a Patient with Aneurysms. Genes. 2022; 13(5):733. https://doi.org/10.3390/genes13050733
Chicago/Turabian StyleWeronska, Anna, Daniel P. Potaczek, Julia Oto, Pilar Medina, Anetta Undas, and Ewa Wypasek. 2022. "A Series of 14 Polish Patients with Thrombotic Events and PC Deficiency-Novel c.401-1G>A PROC Gene Splice Site Mutation in a Patient with Aneurysms" Genes 13, no. 5: 733. https://doi.org/10.3390/genes13050733