
Development and Characterization of Chromosome Segment Substitution Lines Derived from Oryza rufipogon in the Background of the Oryza sativa indica Restorer Line R974

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction and PCR
2.3. Construction of CSSLs
2.4. Evaluation of Salt Stress Tolerance
2.5. QTL Analysis and SNP Chip Assay
3. Results
3.1. Screening of Polymorphic Markers and Linkage Map Construction
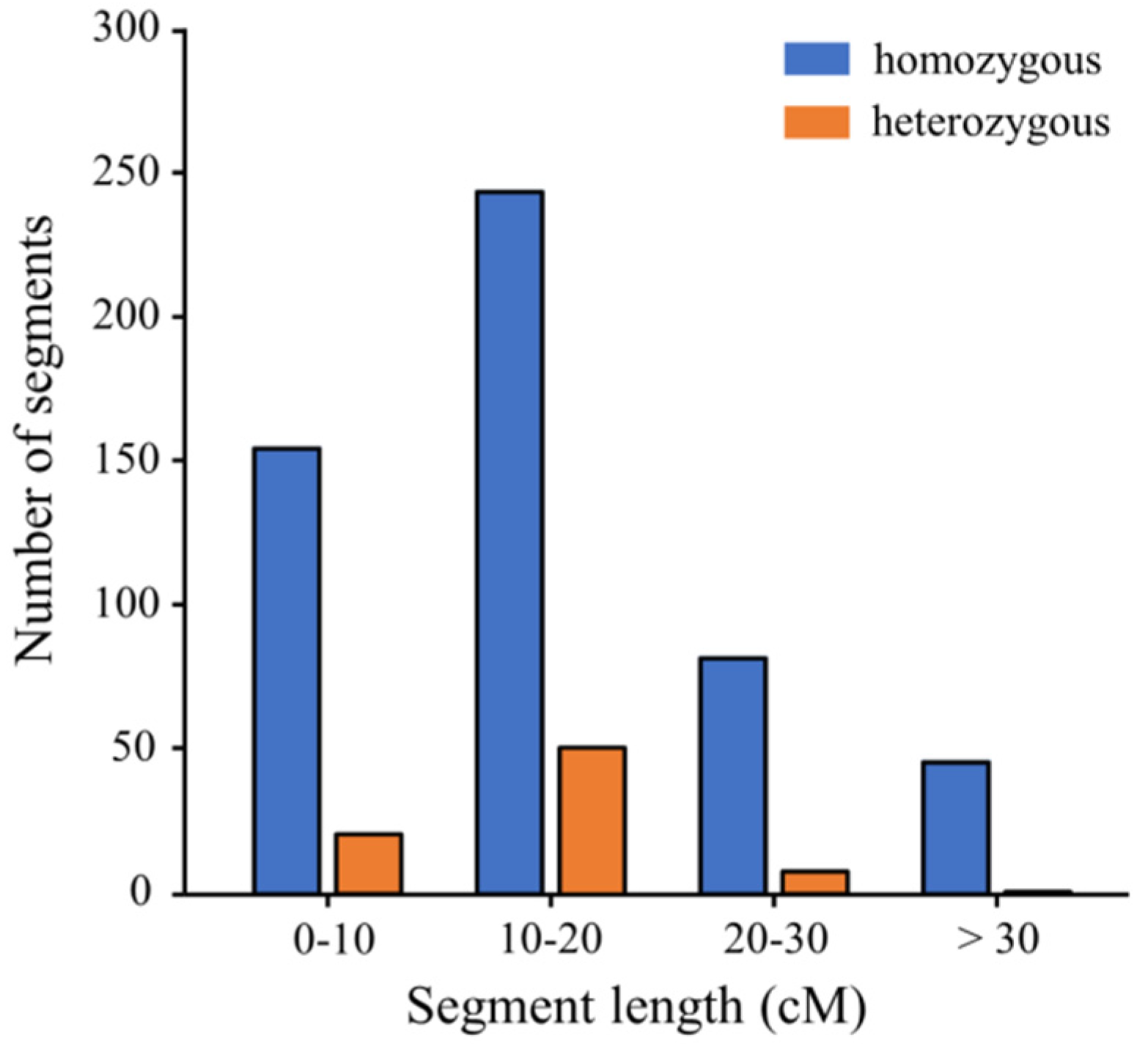
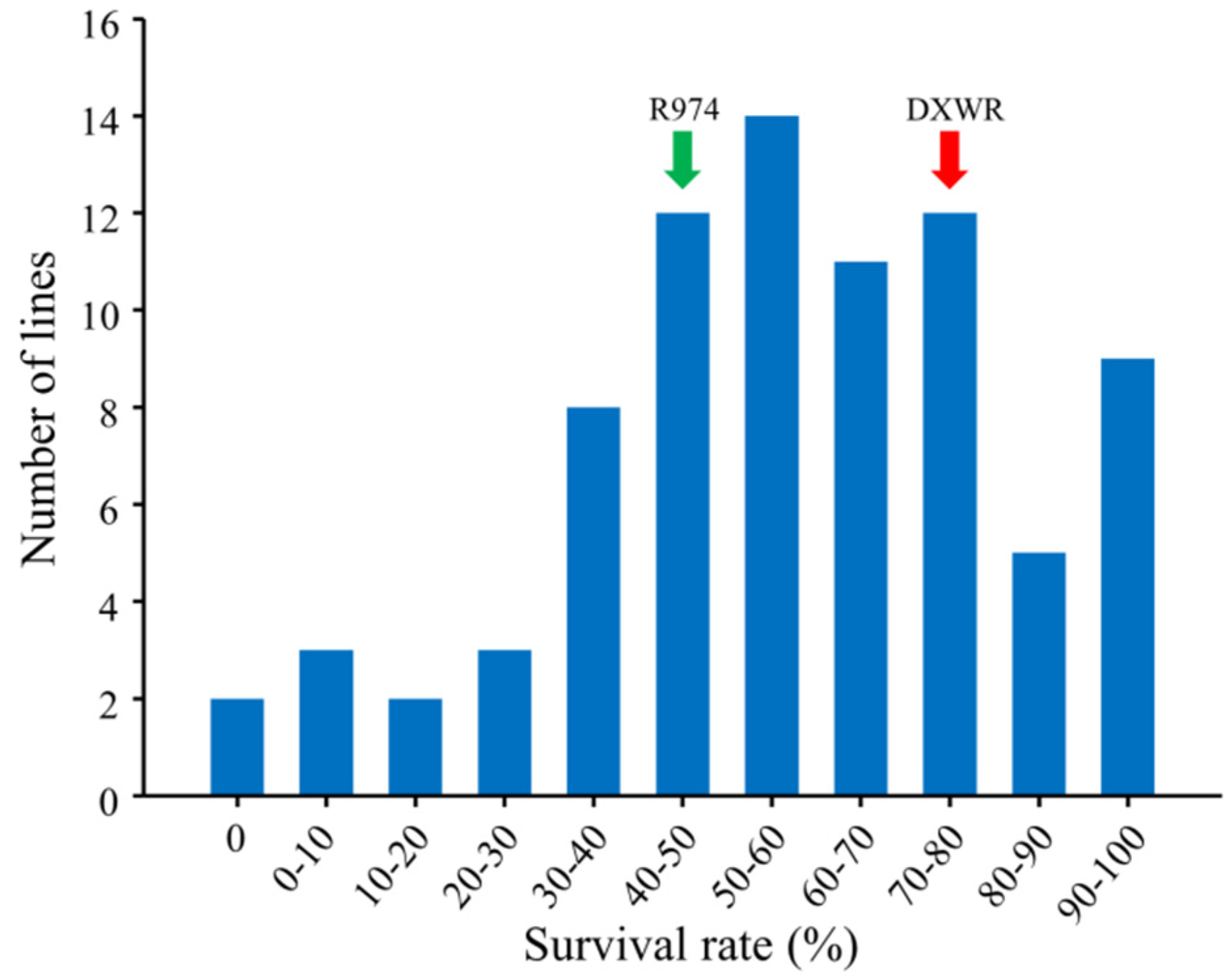
3.2. Development and Characterization of the CSSL Population
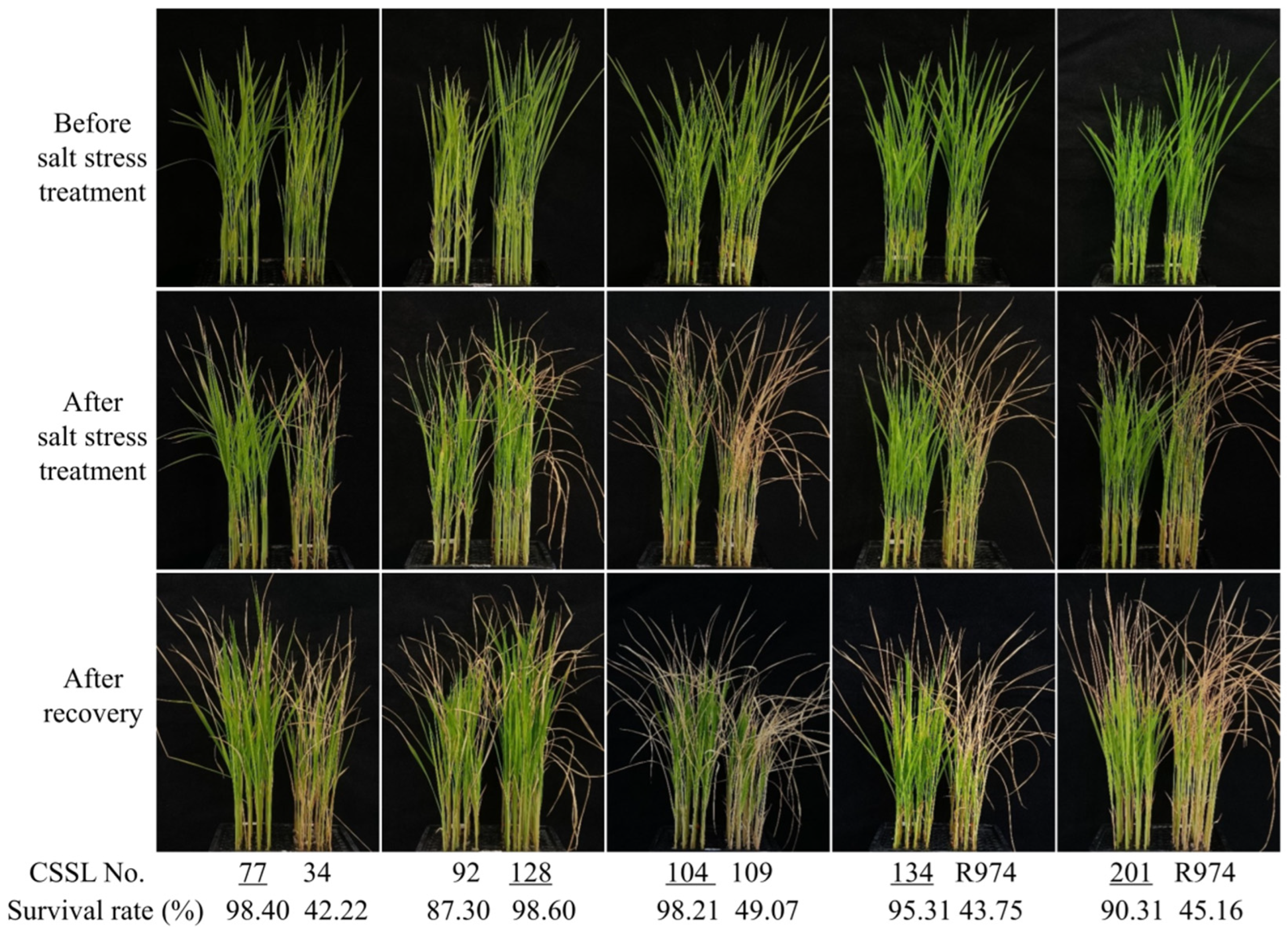
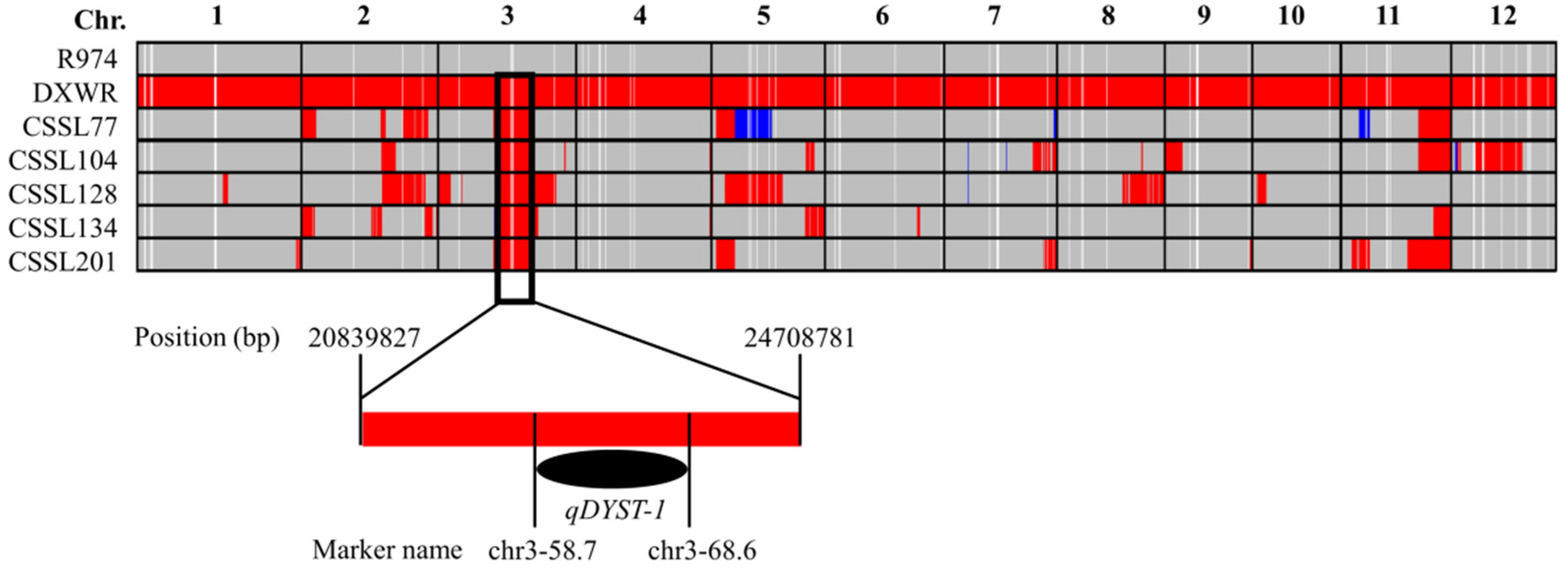
3.3. QTL Mapping for Salt Stress Tolerance
3.4. SNP Chip Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, J.; Wan, L.; Igathinathane, C.; Zhang, Z.; Guo, Y.; Sun, D.; Cen, H. Spatiotemporal heterogeneity of chlorophyll content and fluorescence response within rice (Oryza sativa L.) canopies under different nitrogen treatments. Front. Plant Sci. 2021, 12, 645977. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Long, Y.; Zhao, Z.; Huang, G.; Huang, K.; Zhang, T.; Jiang, Y.; Yuan, Q.; Pei, X. Isolation and characterization of a green-tissue promoter from common wild rice (Oryza rufipogon Griff.). Int. J. Mol. Sci. 2018, 19, 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Shi, J.; Liu, S.; Sun, X.; Huang, J.; Qiao, W.; Cheng, Y.; Zhang, L.; Zheng, X.; Yang, Q. Conservation recommendations for Oryza rufipogon Griff. in China based on genetic diversity analysis. Sci. Rep. 2020, 10, 14375. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Wang, E.; Lin, X.; Ji, L.; Chang, J.; Chen, H.; Wang, J.; Chen, D.; Tran, L.P.; Tian, C. Wild rice harbors more root endophytic fungi than cultivated rice in the F1 offspring after crossbreeding. BMC Genom. 2021, 22, 278. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Fang, Y.; Hu, P.; Tan, Y.; Wang, Y.; Hou, L.; Deng, X.; Wu, H.; Zhu, L.; Zhu, L.; et al. Construction of a high-density genetic map based on SLAF markers and QTL analysis of leaf size in rice. Front. Plant Sci. 2020, 11, 1143. [Google Scholar] [CrossRef]
- Swamy, B.P.; Sarla, N. Yield-enhancing quantitative trait loci (QTLs) from wild species. Biotechnol. Adv. 2008, 26, 106–120. [Google Scholar] [CrossRef]
- Yuan, L.; Zhang, L.; Wei, X.; Wang, R.; Li, N.; Chen, G.; Fan, F.; Huang, S.; Li, J.; Li, S. Quantitative trait locus mapping of salt tolerance in wild rice Oryza longistaminata. Int. J. Mol. Sci. 2022, 23, 2379. [Google Scholar] [CrossRef]
- Qiao, W.; Qi, L.; Cheng, Z.; Su, L.; Li, J.; Sun, Y.; Ren, J.; Zheng, X.; Yang, Q. Development and characterization of chromosome segment substitution lines derived from Oryza rufipogon in the genetic background of O. sativa spp. indica cultivar 9311. BMC Genom. 2016, 17, 580. [Google Scholar]
- Li, Z.K.; Pinson, S.R.M.; Stansel, J.W.; Park, W.D. Identification of quantitative trait loci (QTLs) for heading date and plant height in cultivated rice (Oryza sativa L.). Theor. Appl. Genet. 1995, 91, 374–381. [Google Scholar] [CrossRef]
- Yano, M.; Sasaki, T. Genetic and molecular dissection of quantitative traits in rice. Plant Mol. Biol. 1997, 35, 145–153. [Google Scholar] [CrossRef]
- Tsunematsu, H.; Yoshimura, A.; Harushima, Y.; Nagamura, Y.; Kurata, N.; Yano, M.; Sasaki, T.; Iwata, N. RFLP framework map using recombinant inbred lines in rice. Breed. Sci. 1996, 46, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhao, Q.; Du, P.; Xu, C.; Wang, B.; Feng, Q.; Liu, Q.; Tang, S.; Gu, M.; Han, B.; et al. Developing high throughput genotyped chromosome segment substitution lines based on population whole-genome re-sequencing in rice (Oryza sativa L.). BMC Genom. 2010, 11, 656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, R.; Zhao, N.; Usman, B.; Luo, L.; Liao, S.; Qin, Y.; Nawaz, G.; Li, R. Development of chromosome segment substitution lines (CSSLs) derived from Guangxi wild rice (Oryza rufipogon Griff.) under rice (Oryza sativa L.) background and the identification of QTLs for plant architecture, agronomic traits and cold tolerance. Genes 2020, 11, 980. [Google Scholar] [CrossRef] [PubMed]
- Bessho-Uehara, K.; Furuta, T.; Masuda, K.; Yamada, S.; Angeles-Shim, R.B.; Ashikari, M.; Takashi, T. Construction of rice chromosome segment substitution lines harboring Oryza barthii genome and evaluation of yield-related traits. Breed. Sci. 2017, 67, 408–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, S.; Onogi, A.; Iijima, K.; Hori, K.; Iwata, H.; Yokoyama, W.; Suehiro, M.; Yamasaki, M. Identification of QTLs for rice grain size using a novel set of chromosomal segment substitution lines derived from Yamadanishiki in the genetic background of Koshihikari. Breed. Sci. 2018, 68, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.P.; Xu, Z.S.; Zheng, W.J.; Zhao, W.; Wang, Y.X.; Yu, T.F.; Chen, M.; Zhou, Y.B.; Min, D.H.; Ma, Y.Z.; et al. Genome-wide analysis of the RAV family in soybean and functional identification of GmRAV-03 involvement in salt and drought stresses and exogenous ABA treatment. Front. Plant Sci. 2017, 8, 905. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Kota, S.; Flowers, T.J. Salt tolerance in rice: Seedling and reproductive stage QTL mapping come of age. Theor. Appl. Genet. 2021, 134, 3495–3533. [Google Scholar] [CrossRef]
- Quan, R.; Wang, J.; Hui, J.; Bai, H.; Lyu, X.; Zhu, Y.; Zhang, H.; Zhang, Z.; Li, S.; Huang, R. Improvement of salt tolerance using wild rice genes. Front. Plant Sci. 2018, 8, 2269. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Hu, B.; Fan, Y.; Ding, G.; Yang, W.; Chen, Y.; Chen, Y.; Xie, J.; Zhang, F. Identification, analysis, and confirmation of seed storability-related loci in Dongxiang wild rice (Oryza rufipogon Griff.). Genes 2021, 12, 1831. [Google Scholar] [CrossRef]
- Jiang, W.; Shi, W.; Ma, X.; Zhao, J.; Wang, S.; Tan, L.; Sun, C.; Liu, F. Identification of microRNAs responding to cold stress in Dongxiang common wild rice. Genome 2019, 62, 635–642. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, P.; Cui, F.; Zhang, F.; Luo, X.; Xie, J. Transcriptome analysis of salt stress responsiveness in the seedlings of Dongxiang wild rice (Oryza rufipogon Griff.). PLoS ONE 2016, 11, e0146242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Zhou, Y.; Zhang, M.; Luo, X.; Xie, J. Effects of drought stress on global gene expression profile in leaf and root samples of Dongxiang wild rice (Oryza rufipogon). Biosci. Rep. 2017, 37, BSR20160509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Han, B.; Tang, J.; Zhang, J.; Cui, D.; Geng, L.; Zhou, H.; Li, M.; Han, L. Construction of chromosome segment substitution lines of Dongxiang common wild rice (Oryza rufipogon Griff.) in the background of the japonica rice cultivar Nipponbare (Oryza sativa L.). Plant Physiol. Biochem. 2019, 144, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tian, Q.; Zhou, S.; Mao, D.; Chen, L. A quantitative proteomic analysis of the molecular mechanism underlying fertility conversion in thermo-sensitive genetic male sterility line AnnongS-1. BMC Plant. Biol. 2019, 19, 65. [Google Scholar] [CrossRef]
- Xie, J.; Agrama, H.A.; Kong, D.; Zhuang, J.; Hu, B.L.; Wan, Y.; Yan, W.G. Genetic diversity associated with conservation of endangered Dongxiang wild rice (Oryza rufipogon). Genet. Resour. Crop Evol. 2010, 57, 597–609. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 5. [Google Scholar]
- Liang, Q.; Wen, D.; Xie, J.; Liu, L.; Wei, Y.; Wang, Y.; Shi, S. A rapid and effective method for silver staining of PCR products separated in polyacrylamide gels. Electrophoresis 2014, 35, 2520–2523. [Google Scholar] [CrossRef]
- Lander, E.S.; Green, P.; Abrahamson, J.; Barlow, A.; Daly, M.J.; Lincoln, S.E.; Newburg, L. MAPMAKER: An interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1987, 1, 174–181. [Google Scholar] [CrossRef]
- Surapaneni, M.; Balakrishnan, D.; Mesapogu, S.; Addanki, K.R.; Yadavalli, V.R.; Tripura Venkata, V.G.N.; Neelamraju, S. Identification of major effect QTLs for agronomic traits and CSSLs in rice from Swarna/Oryza nivara derived backcross inbred lines. Front. Plant Sci. 2017, 8, 1027. [Google Scholar] [CrossRef]
- Van Berloo, R. GGT 2.0: Versatile software for visualization and analysis of genetic data. J. Hered. 2008, 99, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Li, H.H.; Ribaut, J.M.; Li, Z.L.; Wang, J.K. Inclusive composite interval mapping (ICIM) for digenic epistasis of quantitative traits in biparental populations. Theor. Appl. Genet. 2008, 116, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Xiao, X.; Gong, J.; Li, P.; Zhao, Y.; Feng, J.; Peng, R.; Shi, Y.; Yuan, Y. Identification of candidate cotton genes associated with fiber length through quantitative trait loci mapping and RNA-sequencing using a chromosome segment substitution line. Front. Plant Sci. 2021, 12, 796722. [Google Scholar] [CrossRef]
- He, J.; Zhang, D.; Chen, X.; Li, Y.; Hu, M.; Sun, S.; Su, Q.; Su, Y.; Li, S. Identification of QTLs and a candidate gene for reducing pre-harvest sprouting in Aegilops tauschii-Triticum aestivum chromosome segment substitution lines. Int. J. Mol. Sci. 2021, 22, 3729. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Qin, R.; Li, C.; Liu, C.; Jiang, Y.; Yu, J.; Chang, D.; Roberts, P.A.; Chen, Q.; Wang, C. Transgressive resistance to Heterodera glycines in chromosome segment substitution lines derived from susceptible soybean parents. Plant Genome 2021, 14, e20091. [Google Scholar] [CrossRef]
- Balakrishnan, D.; Surapaneni, M.; Mesapogu, S.; Neelamraju, S. Development and use of chromosome segment substitution lines as a genetic resource for crop improvement. Theor. Appl. Genet. 2019, 132, 1–25. [Google Scholar] [CrossRef]
- Alyr, M.H.; Pallu, J.; Sambou, A.; Nguepjop, J.R.; Seye, M.; Tossim, H.A.; Djiboune, Y.R.; Sane, D.; Rami, J.F.; Fonceka, D. Fine-mapping of a wild genomic region involved in pod and seed size reduction on chromosome A07 in peanut (Arachis hypogaea L.). Genes 2020, 11, 1402. [Google Scholar] [CrossRef]
- Yuan, L.P.; Virmani, S.S.; Mao, C.X. Hybrid rice: Achievements and Further Outlook; International Rice Research Institute: Manila, Philippines, 1989; pp. 219–223. [Google Scholar]
- Yuan, L.P. Advantages of and constraints to the use of hybrid rice varieties. In International Workshop on Apomixis in Rice; Wilson, K.J., Ed.; Hunan Hybrid Rice Research Center: Changsha, China, 1993. [Google Scholar]
- Zaid, I.U.; Tang, W.; Liu, E.; Khan, S.U.; Wang, H.; Mawuli, E.W.; Hong, D. Genome-wide single-nucleotide polymorphisms in CMS and restorer lines discovered by genotyping using sequencing and association with marker-combining ability for 12 yield-related traits in Oryza sativa L. subsp. Japonica. Front. Plant Sci. 2017, 8, 143. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.S.; Liu, Q.L.; He, F.Q.; Gu, X.H.; Liao, Y.F. Breeding of early indica good quality restorer line T0974 and its application. Hybrid Rice 1997, 12, 1–3. [Google Scholar]
- Kakar, N.; Jumaa, S.H.; Redoña, E.D.; Warburton, M.L.; Reddy, K.R. Evaluating rice for salinity using pot-culture provides a systematic tolerance assessment at the seedling stage. Rice 2019, 12, 57. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Jiang, H.; Meng, L.; Chen, J. Gene mapping, cloning and association analysis for salt tolerance in rice. Int. J. Mol. Sci. 2021, 22, 11674. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, W.; Zhang, F.; Luo, X.; Hu, B.; Xie, J. Metabolomic profiling of Dongxiang wild rice under salinity demonstrates the significant role of amino acids in rice salt stress. Front. Plant Sci. 2021, 12, 729004. [Google Scholar] [CrossRef]
- Kitony, J.K.; Sunohara, H.; Tasaki, M.; Mori, J.I.; Shimazu, A.; Reyes, V.P.; Yasui, H.; Yamagata, Y.; Yoshimura, A.; Yamasaki, M.; et al. Development of an aus-derived nested association mapping (Aus-NAM) population in rice. Plants 2021, 10, 1255. [Google Scholar] [CrossRef]
- Reyes, V.P.; Angeles-Shim, R.B.; Mendioro, M.S.; Manuel, M.C.C.; Lapis, R.S.; Shim, J.; Sunohara, H.; Nishiuchi, S.; Kikuta, M.; Makihara, D.; et al. Marker-assisted introgression and stacking of major QTLs controlling grain number (Gn1a) and number of primary branching (WFP) to NERICA cultivars. Plants 2021, 10, 844. [Google Scholar] [CrossRef]
- Schuster, C.F.; Wiedemann, D.M.; Kirsebom, F.C.M.; Santiago, M.; Walker, S.; Gründling, A. High-throughput transposon sequencing highlights the cell wall as an important barrier for osmotic stress in methicillin resistant Staphylococcus aureus and underlines a tailored response to different osmotic stressors. Mol. Microbiol. 2020, 113, 699–717. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Cao, Y.; Wang, Z.; Wang, Z.Q.; Shi, J.; Liang, X.; Song, W.; Chen, Q.; Lai, J.; Jiang, C. A retrotransposon in an HKT1 family sodium transporter causes variation of leaf Na+ exclusion and salt tolerance in maize. New Phytol. 2018, 217, 1161–1176. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.R.; Yang, K.; Wang, X.; Lin, X.L.; Rui, L.; Liu, H.F.; Liu, D.D.; You, C.X. Overexpression of MdZAT5, an C2H2-Type zinc finger protein, regulates anthocyanin accumulation and salt stress response in Apple Calli and Arabidopsis. Int. J. Mol. Sci. 2022, 23, 1897. [Google Scholar] [CrossRef]
- Yu, Z.; Yan, H.; Liang, L.; Zhang, Y.; Yang, H.; Li, W.; Choi, J.; Huang, J.; Deng, S. A C2H2-type zinc-finger protein from Millettia pinnata, MpZFP1, enhances salt tolerance in transgenic Arabidopsis. Int. J. Mol. Sci. 2021, 22, 10832. [Google Scholar] [CrossRef]
- Fu, M.; Kang, H.K.; Son, S.H.; Kim, S.K.; Nam, K.H. A subset of Arabidopsis RAV transcription factors modulates drought and salt stress responses independent of ABA. Plant Cell Physiol. 2014, 55, 1892–1904. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr. | Chr. Length (cM) | No. of Markers | Average Distance between Adjacent Markers (cM) |
|---|---|---|---|
| 1 | 229.70 | 20 | 11.49 |
| 2 | 160.30 | 12 | 13.36 |
| 3 | 128.70 | 15 | 8.58 |
| 4 | 117.70 | 11 | 10.70 |
| 5 | 128.20 | 9 | 14.24 |
| 6 | 137.20 | 11 | 12.47 |
| 7 | 71.30 | 11 | 6.48 |
| 8 | 119.50 | 12 | 9.96 |
| 9 | 115.30 | 11 | 10.48 |
| 10 | 105.90 | 9 | 11.77 |
| 11 | 87.80 | 8 | 10.98 |
| 12 | 154.70 | 11 | 14.06 |
| Total | 1556.30 | 140 | 11.12 |
| Chr. | Homozygous Segments | Heterozygous Segments | Total Segment Length (cM) | Effective Coverage Length (cM) (Homo) | Genome Coverage (%) (Homo) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Number of Segments | Total Segment Length (cM) | Average Length (cM) | Number of Segments | Total Segment Length (cM) | Average Length (cM) | ||||
| 1 | 70 | 1104.40 | 15.78 | 12 | 165.70 | 13.81 | 1270.10 | 222.60 | 96.91 |
| 2 | 64 | 1018.60 | 15.92 | 8 | 139.05 | 17.38 | 1157.65 | 160.30 | 100.00 |
| 3 | 55 | 688.75 | 12.52 | 5 | 53.00 | 10.60 | 741.75 | 117.15 | 91.03 |
| 4 | 36 | 569.30 | 15.81 | 4 | 49.95 | 12.49 | 619.25 | 105.00 | 89.21 |
| 5 | 47 | 885.05 | 18.83 | 10 | 170.70 | 17.07 | 1055.75 | 101.70 | 79.33 |
| 6 | 42 | 769.55 | 18.32 | 9 | 143.35 | 15.93 | 912.90 | 107.05 | 78.02 |
| 7 | 24 | 233.80 | 9.74 | 1 | 10.05 | 10.05 | 243.85 | 56.05 | 78.61 |
| 8 | 45 | 715.60 | 15.90 | 10 | 117.85 | 11.79 | 833.45 | 119.50 | 100.00 |
| 9 | 28 | 554.45 | 19.80 | 3 | 23.75 | 7.92 | 578.20 | 115.30 | 100.00 |
| 10 | 35 | 631.90 | 18.05 | 8 | 113.50 | 14.19 | 745.40 | 105.90 | 100.00 |
| 11 | 32 | 518.40 | 16.20 | 2 | 29.35 | 14.68 | 547.75 | 87.80 | 100.00 |
| 12 | 49 | 993.70 | 20.28 | 9 | 92.65 | 10.29 | 1086.35 | 154.70 | 100.00 |
| Average | 43.92 | 723.63 | 6.75 | 92.41 | 92.76 | ||||
| Total | 527 | 8683.50 | 16.48 | 81 | 1108.90 | 13.69 | 9792.40 | 1453.05 | 93.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, G.; Hu, B.; Zhou, Y.; Yang, W.; Zhao, M.; Xie, J.; Zhang, F. Development and Characterization of Chromosome Segment Substitution Lines Derived from Oryza rufipogon in the Background of the Oryza sativa indica Restorer Line R974. Genes 2022, 13, 735. https://doi.org/10.3390/genes13050735
Ding G, Hu B, Zhou Y, Yang W, Zhao M, Xie J, Zhang F. Development and Characterization of Chromosome Segment Substitution Lines Derived from Oryza rufipogon in the Background of the Oryza sativa indica Restorer Line R974. Genes. 2022; 13(5):735. https://doi.org/10.3390/genes13050735
Chicago/Turabian StyleDing, Gumu, Biaolin Hu, Yi Zhou, Wanling Yang, Minmin Zhao, Jiankun Xie, and Fantao Zhang. 2022. "Development and Characterization of Chromosome Segment Substitution Lines Derived from Oryza rufipogon in the Background of the Oryza sativa indica Restorer Line R974" Genes 13, no. 5: 735. https://doi.org/10.3390/genes13050735