
An Alternatively Spliced Variant of METTL3 Mediates Tumor Suppression in Hepatocellular Carcinoma


Abstract
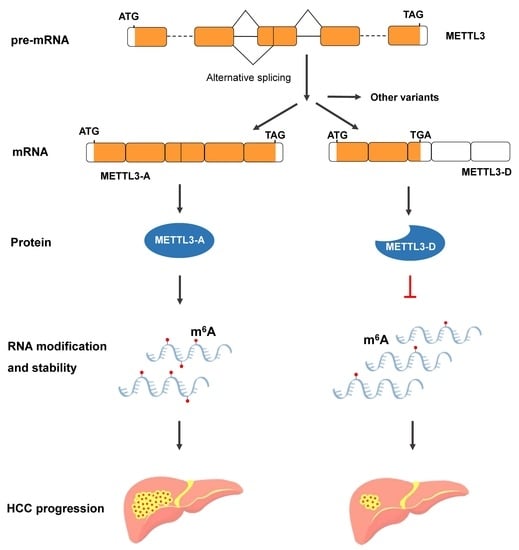
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Human Specimens
2.3. Plasmid Transfection and Antibodies
2.4. PCR and Sequencing
2.5. Quantitative Real-Time PCR
2.6. Bioinformatics and Online Resource
2.7. RNA m6A Quantification
2.8. Cell Growth and Proliferation Assays
2.9. Cell Migration and Invasion Assays
2.10. Statistical Analysis
3. Results
3.1. Differentially Spliced Variants of METTL3 in Human Tissues and HCC Cell Lines
3.2. METTL3-D Is Negatively Associated with HCC Malignancy
3.3. Expression of METTL3-D Decreases m6A Modification
3.4. METTL3-D Levels Are Inversely Correlated with Patient Survival in Multiple Cancers
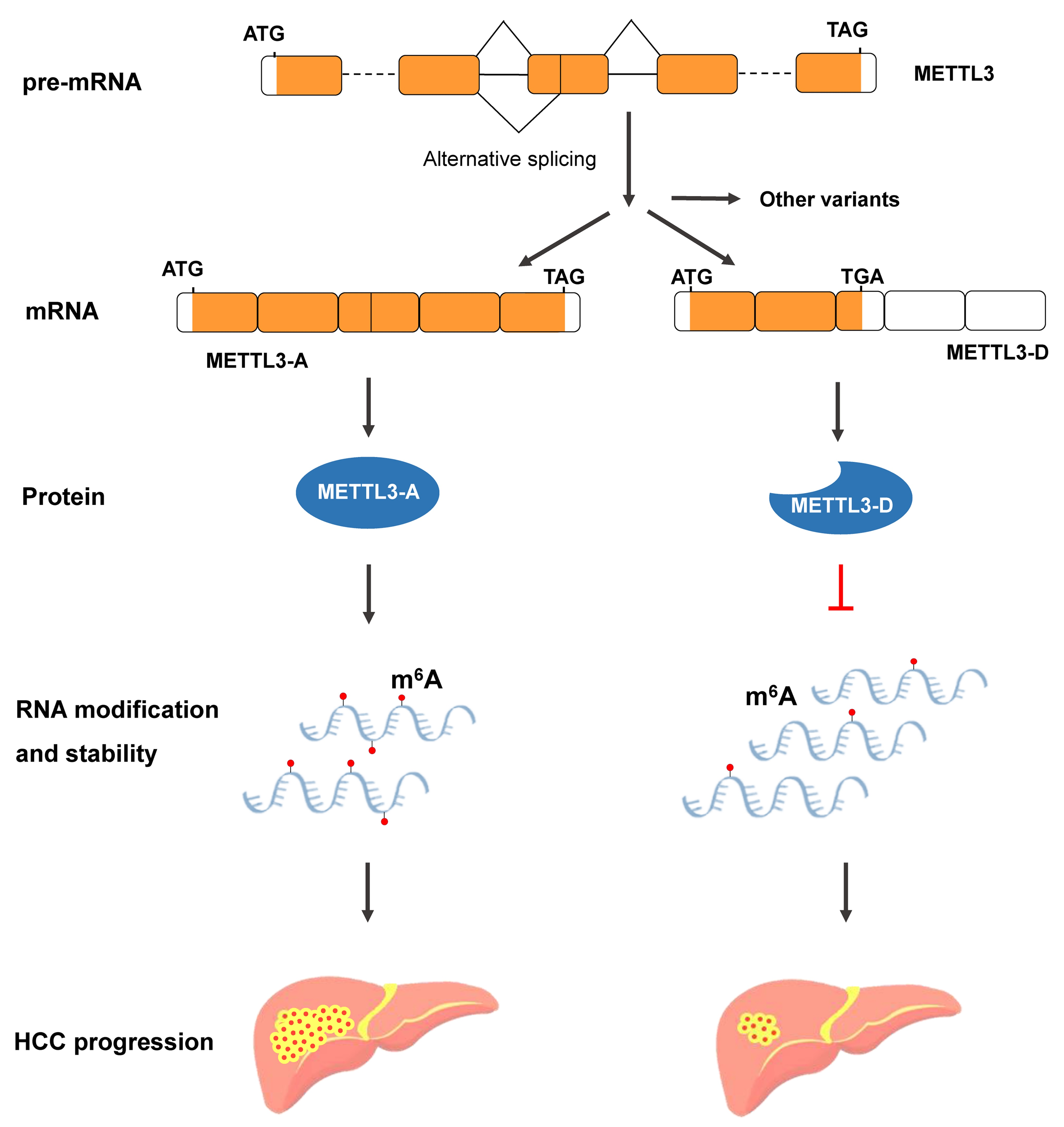
3.5. METTL3-D Inhibits the Proliferation, Migration, and Invasion of HCC Cells
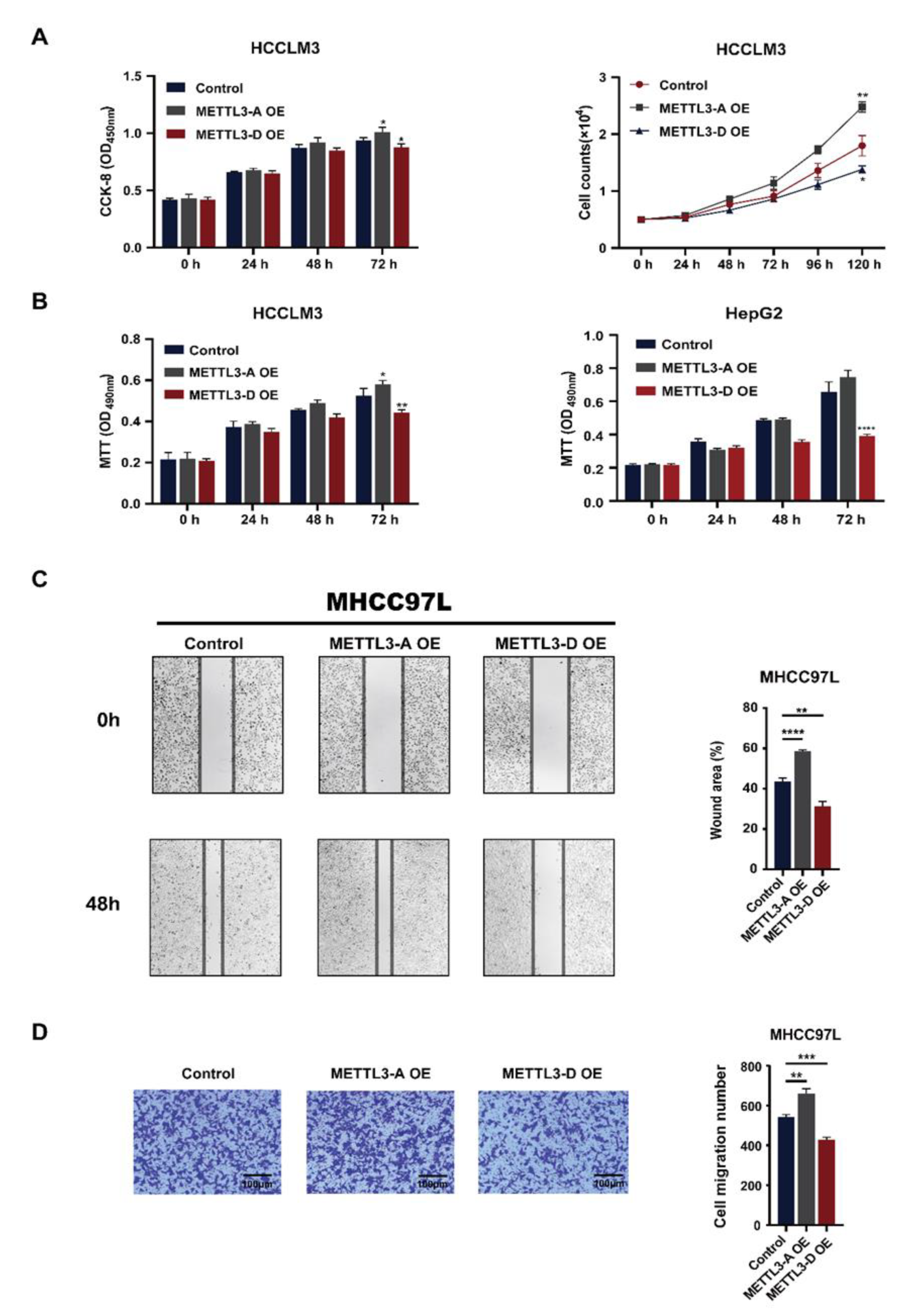
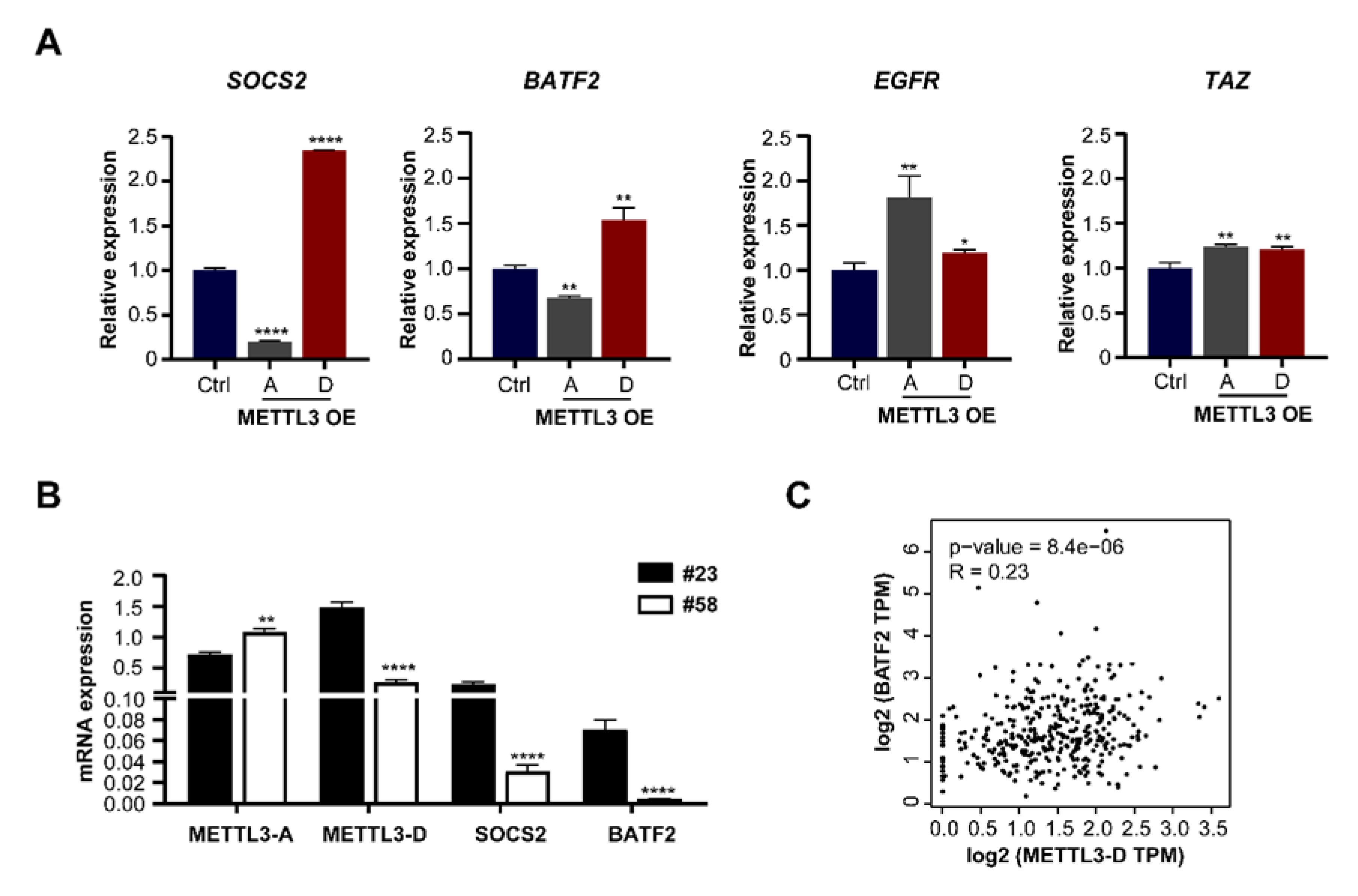
3.6. METTL3-D Has an Opposite Function to Downstream Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharp, P.A. The centrality of RNA. Cell 2009, 136, 577–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Zink, D.; Korn, B.; Vingron, M.; Haas, S.A. Genome wide identification and classification of alternative splicing based on EST data. Bioinformatics 2004, 20, 2579–2585. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamm, S.; Ben-Ari, S.; Rafalska, I.; Tang, Y.; Zhang, Z.; Toiber, D.; Thanaraj, T.; Soreq, H. Function of alternative splicing. Gene 2005, 344, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bao, Y.; Zhang, S.; Wang, Z. Splicing dysregulation in cancer: From mechanistic understanding to a new class of therapeutic targets. Sci. China Life Sci. 2020, 63, 469–484. [Google Scholar] [CrossRef]
- Huun, J.; Gansmo, L.B.; Mannsåker, B.; Iversen, G.T.; Øvrebø, J.I.; Lønning, P.E.; Knappskog, S. Impact of the MDM2 splice-variants MDM2-A, MDM2-B and MDM2-C on cytotoxic stress response in breast cancer cells. BMC Cell Biol. 2017, 18, 17. [Google Scholar] [CrossRef]
- Mercatante, D.R.; Mohler, J.L.; Kole, R. Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J. Biol. Chem. 2002, 277, 49374–49382. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.-T.; Ma, W.K.; Scharner, J.; Liu, Y.-R.; Krainer, A.R. A human-specific switch of alternatively spliced AFMID isoforms contributes to TP53 mutations and tumor recurrence in hepatocellular carcinoma. Genome Res. 2018, 28, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Yu, J.; Han, W.; Fan, X.; Qian, H.; Wei, H.; Yi-hsuan, S.T.; Zhao, J.; Zhang, W.; Liu, Q. A splicing isoform of TEAD4 attenuates the Hippo–YAP signalling to inhibit tumour proliferation. Nat. Commun. 2016, 7, ncomms11840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Hu, Z.; Zhao, Y.; Huang, S.; He, X. Transcriptome-wide analysis reveals the landscape of aberrant alternative splicing events in liver cancer. Hepatology 2019, 69, 359–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.-Q.; Zhou, Y.-J.; Qiu, L.-X.; Wang, B.; Yang, Y.; Liao, W.-T.; Luo, Y.-H.; Shi, Y.-H.; Zhou, J.; Fan, J. Prognostic alternative mRNA splicing signature in hepatocellular carcinoma: A study based on large-scale sequencing data. Carcinogenesis 2019, 40, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, X.; Zhang, X.; Chen, R. Identification of important long non-coding RNAs and highly recurrent aberrant alternative splicing events in hepatocellular carcinoma through integrative analysis of multiple RNA-Seq datasets. Mol. Genet. Genom. 2016, 291, 1035–1051. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Wang, C.; Jin, G.; Ruan, H.; Gu, D.; Wei, L.; Wang, H.; Wang, N.; Arunachalam, E.; Zhang, Y. Regulator of calcineurin 1 gene isoform 4, down-regulated in hepatocellular carcinoma, prevents proliferation, migration, and invasive activity of cancer cells and metastasis of orthotopic tumors by inhibiting nuclear translocation of NFAT1. Gastroenterology 2017, 153, 799–811.e33. [Google Scholar] [CrossRef] [Green Version]
- Snider, N.T.; Altshuler, P.J.; Wan, S.; Welling, T.H.; Cavalcoli, J.; Omary, M.B. Alternative splicing of human NT5E in cirrhosis and hepatocellular carcinoma produces a negative regulator of ecto-5′-nucleotidase (CD73). Mol. Biol. Cell 2014, 25, 4024–4033. [Google Scholar] [CrossRef]
- Wang, Z.; Li, S.S.-C. Numb: A new player in EMT. Cell Adhes. Migr. 2010, 4, 176–179. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Xu, W.; Ji, J.; Feng, D.; Sourbier, C.; Yang, Y.; Qu, J.; Zeng, Z.; Wang, C.; Chang, X. Alternative splicing of the cell fate determinant Numb in hepatocellular carcinoma. Hepatology 2015, 62, 1122–1131. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hsu, P.J.; Chen, Y.-S.; Yang, Y.-G. Dynamic transcriptomic m6A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M. Topology of the human and mouse m6A RNA methylomes revealed by m 6 A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- He, P.C.; He, C. m6A RNA methylation: From mechanisms to therapeutic potential. EMBO J. 2021, 40, e105977. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.J.; Zhu, Y.; Ma, H.; Guo, Y.; Shi, X.; Liu, Y.; Qi, M.; Lu, Z.; Shi, H.; Wang, J. Ythdc2 is an N 6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017, 27, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zang, L.; Zhang, F.; Chen, J.; Shen, H.; Shu, L.; Liang, F.; Feng, C.; Chen, D.; Tao, H. Fat mass and obesity-associated (FTO) protein regulates adult neurogenesis. Hum. Mol. Genet. 2017, 26, 2398–2411. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, M.; Zhao, Y.-L.; Yang, Y.; Yang, Y.-G. RNA methylations in human cancers. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Bokar, J.A.; Rath-Shambaugh, M.E.; Ludwiczak, R.; Narayan, P.; Rottman, F. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J. Biol. Chem. 1994, 269, 17697–17704. [Google Scholar] [CrossRef]
- Bujnicki, J.M.; Feder, M.; Radlinska, M.; Blumenthal, R.M. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA: m6A methyltransferase. J. Mol. Evol. 2002, 55, 431–444. [Google Scholar] [CrossRef]
- Bokar, J.; Shambaugh, M.; Polayes, D.; Matera, A.; Rottman, F. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar]
- Batista, P.J.; Molinie, B.; Wang, J.; Qu, K.; Zhang, J.; Li, L.; Bouley, D.M.; Lujan, E.; Haddad, B.; Daneshvar, K. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 2014, 15, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N 6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.C.; Shen, J.; Cheng, C.L.H.; Tsang, L.H.; Ho, D.W.H.; Chiu, D.K.C.; Lee, J.M.F. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef]
- Xie, J.-W.; Huang, X.-B.; Chen, Q.-Y.; Ma, Y.-B.; Zhao, Y.-J.; Liu, L.-C.; Wang, J.-B.; Lin, J.-X.; Lu, J.; Cao, L.-L. m6A modification-mediated BATF2 acts as a tumor suppressor in gastric cancer through inhibition of ERK signaling. Mol. Cancer 2020, 19, 114. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, X.-L.; Sun, B.-F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.-J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.-S. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N 6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wong, C.-M. The emerging roles of N6-methyladenosine (m6A) deregulation in liver carcinogenesis. Mol. Cancer 2020, 19, 44. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yin, Z.; Hou, B.; Yu, M.; Chen, R.; Jin, H.; Jian, Z. Expression profiles and prognostic significance of RNA N6-methyladenosine-related genes in patients with hepatocellular carcinoma: Evidence from independent datasets. Cancer Manag. Res. 2019, 11, 3921. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751. [Google Scholar] [CrossRef] [Green Version]
- Buels, R.; Yao, E.; Diesh, C.M.; Hayes, R.D.; Munoz-Torres, M.; Helt, G.; Goodstein, D.M.; Elsik, C.G.; Lewis, S.E.; Stein, L. JBrowse: A dynamic web platform for genome visualization and analysis. Genome Biol. 2016, 17, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasband, W. WS 1997-2018. ImageJ; US National Institutes of Health: Bethesda, MD, USA, 2018. [Google Scholar]
- Pan, F.; Lin, X.; Hao, L.; Chu, X.; Wan, H.; Wang, R. The role of RNA methyltransferase METTL3 in hepatocellular carcinoma: Results and perspectives. Front. Cell Dev. Biol. 2021, 9, 1191. [Google Scholar] [CrossRef]
- Liu, L.; Wu, Y.; Li, Q.; Liang, J.; He, Q.; Zhao, L.; Chen, J.; Cheng, M.; Huang, Z.; Ren, H. METTL3 Promotes Tumorigenesis and Metastasis through BMI1 m6A Methylation in Oral Squamous Cell Carcinoma. Mol. Ther. 2020, 28, 2177–2190. [Google Scholar] [CrossRef]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef]
- Supek, F.; Lehner, B.; Lindeboom, R.G. To NMD or not to NMD: Nonsense-mediated mRNA decay in cancer and other genetic diseases. Trends Genet. 2020, 37, 657–668. [Google Scholar] [CrossRef]
- Pawlicka, K.; Kalathiya, U.; Alfaro, J. Nonsense-mediated mRNA decay: Pathologies and the potential for novel therapeutics. Cancers 2020, 12, 765. [Google Scholar] [CrossRef] [Green Version]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef]
- Sen, S. Aberrant pre-mRNA splicing regulation in the development of hepatocellular carcinoma. Hepatoma Res. 2018, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Xie, S.; Zhang, C.; Zhu, F. Aberrant regulation of alternative pre-mRNA splicing in hepatocellular carcinoma. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Alcedo, K.P.; Kim, H.J.; Snider, N.T. Alternative splicing in hepatocellular carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Luo, D.; Singh, R.N. Pre-mRNA splicing modulation by antisense oligonucleotides. In Exon Skipping and Inclusion Therapies; Springer: New York, NY, USA, 2018; pp. 415–437. [Google Scholar]
- Lim, K.H.; Han, Z.; Jeon, H.Y.; Kach, J.; Jing, E.; Weyn-Vanhentenryck, S.; Downs, M.; Corrionero, A.; Oh, R.; Scharner, J. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun. 2020, 11, 3501. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, R.-Y.; Ding, Z.; Zhao, Q.; Ke, T.-Y.; Chen, S.; Wang, X.-Y.; Wang, Y.-Y.; Sheng, M.-F.; Wang, W.; Long, N.; et al. An Alternatively Spliced Variant of METTL3 Mediates Tumor Suppression in Hepatocellular Carcinoma. Genes 2022, 13, 669. https://doi.org/10.3390/genes13040669
Xu R-Y, Ding Z, Zhao Q, Ke T-Y, Chen S, Wang X-Y, Wang Y-Y, Sheng M-F, Wang W, Long N, et al. An Alternatively Spliced Variant of METTL3 Mediates Tumor Suppression in Hepatocellular Carcinoma. Genes. 2022; 13(4):669. https://doi.org/10.3390/genes13040669
Chicago/Turabian StyleXu, Rui-Yao, Zhan Ding, Qing Zhao, Tiao-Ying Ke, Shu Chen, Xing-Yu Wang, Yao-Yun Wang, Meng-Fei Sheng, Wei Wang, Ni Long, and et al. 2022. "An Alternatively Spliced Variant of METTL3 Mediates Tumor Suppression in Hepatocellular Carcinoma" Genes 13, no. 4: 669. https://doi.org/10.3390/genes13040669