
Breast Tumour Kinase (Brk/PTK6) Contributes to Breast Tumour Xenograft Growth and Modulates Chemotherapeutic Responses In Vitro

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Interference
2.3. Human Tumour Xenografts
2.4. Dose Response Determinations
2.5. Western Blot
2.6. Human Breast Tumour Samples
2.7. Tissue Processing, RNA Extraction, and cDNA Synthesis
2.8. Real-Time PCR
2.9. Statistical Analysis
3. Results
3.1. Brk Promotes Breast Xenograft Formation
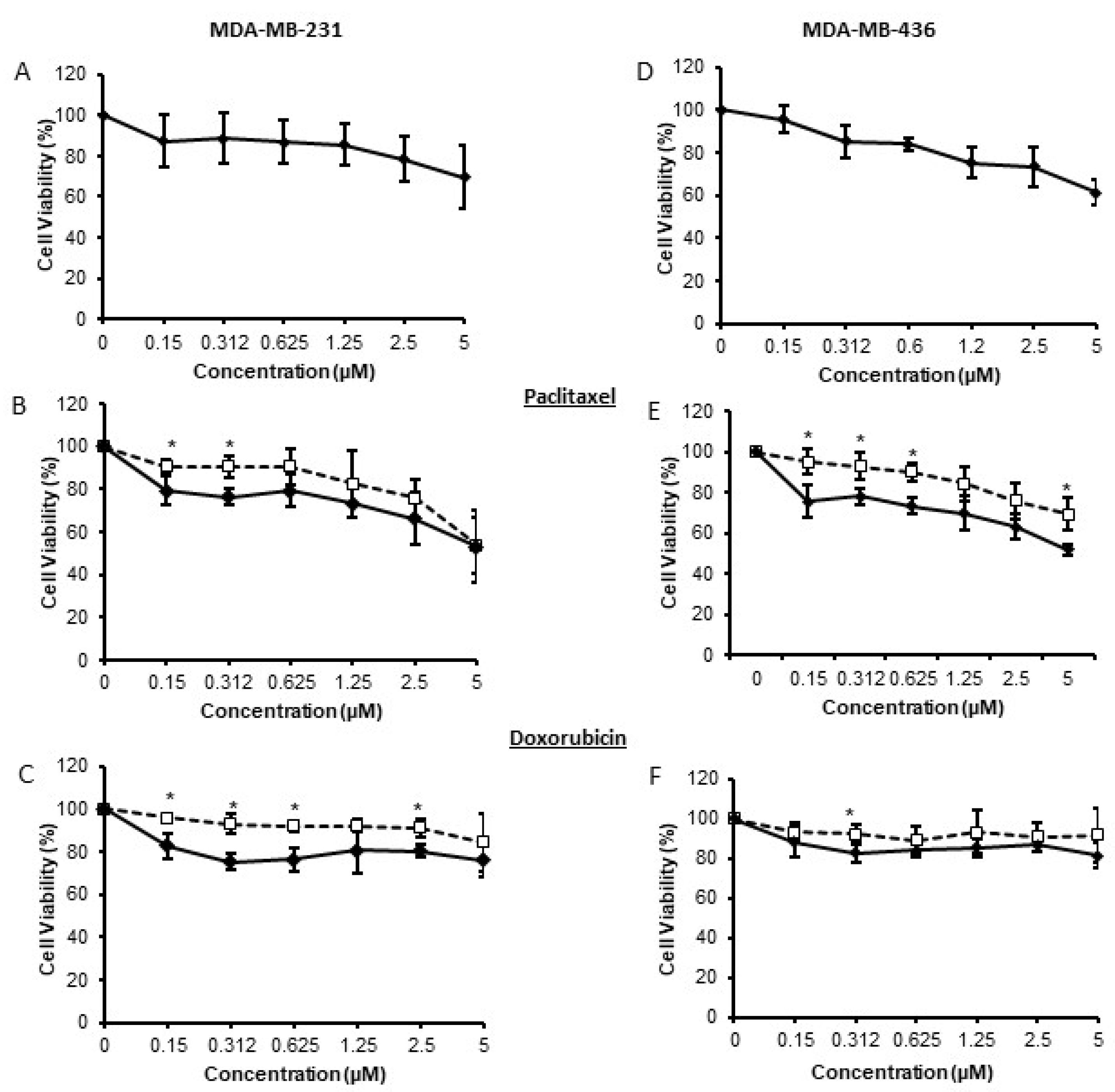
3.2. Brk Reduces Breast Cancer Cell Susceptibility to Chemotherapeutic Agents In Vitro
3.3. Brk Inhibition in Triple Negative Breast Cancers
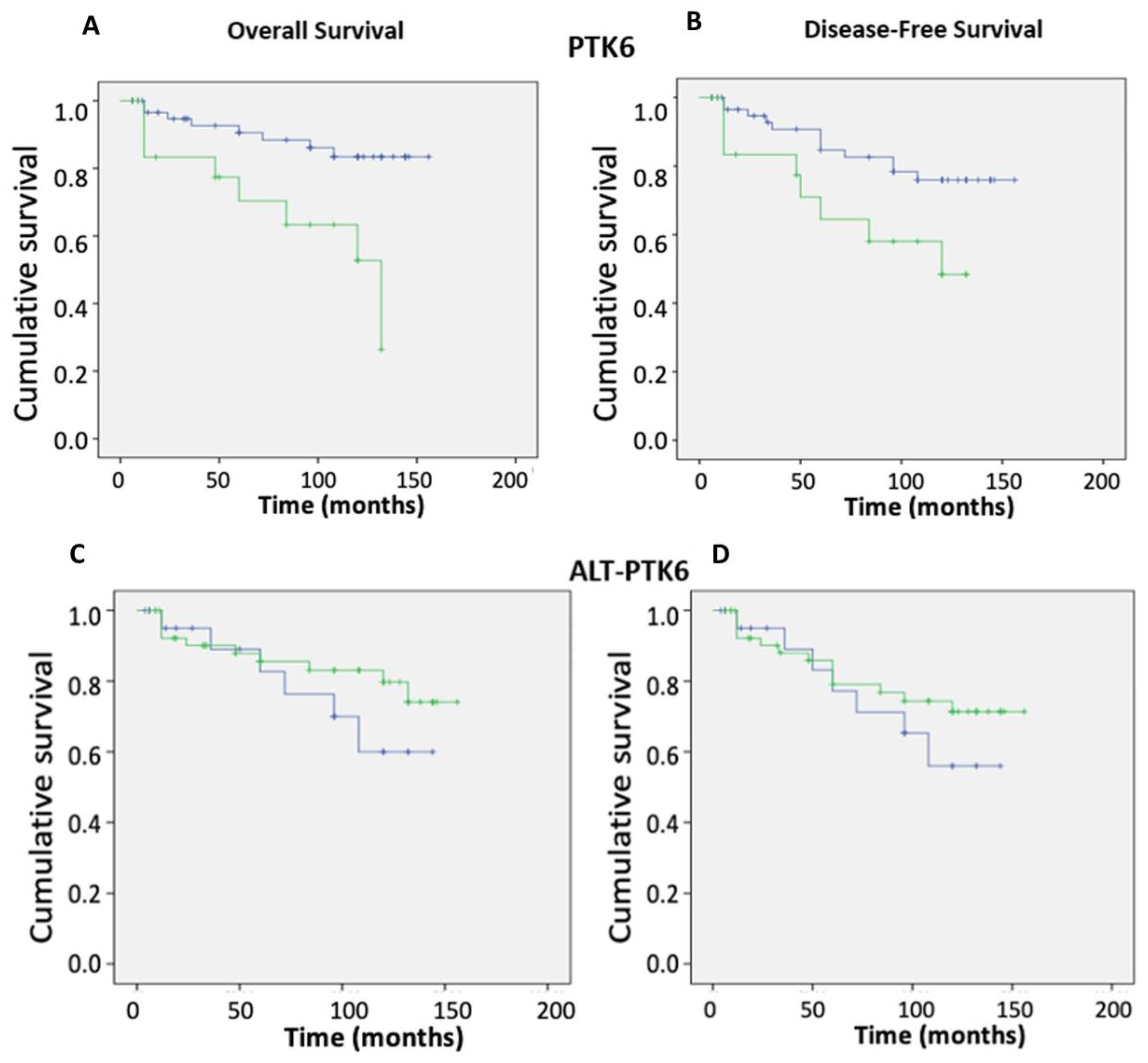
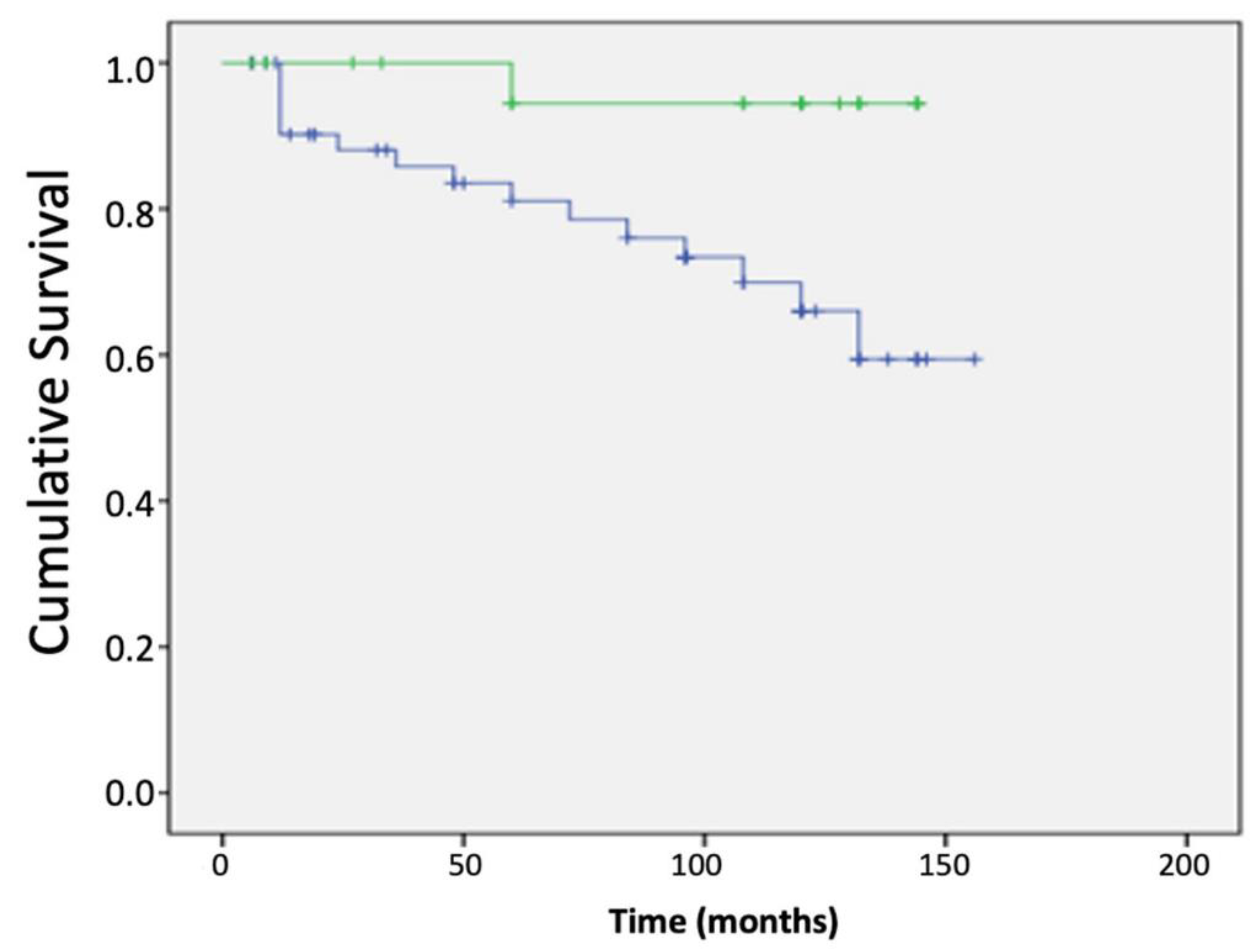
3.4. Impact of Brk Splice Variants on Patient Outcomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harvey, A.J.; Pennington, C.J.; Porter, S.; Burmi, R.S.; Edwards, D.R.; Court, W.; Eccles, S.A.; Crompton, M.R. Brk protects breast cancer cells from autophagic cell death induced by loss of anchorage. Am. J. Pathol. 2009, 175, 1226–1234. [Google Scholar] [CrossRef]
- Ostrander, J.H.; Daniel, A.R.; Lofgren, K.; Kleer, C.G.; Lange, C.A. Breast tumor kinase (protein tyrosine kinase 6) regulates heregulin-induced activation of erk5 and p38 map kinases in breast cancer cells. Cancer Res. 2007, 67, 4199–4209. [Google Scholar] [CrossRef] [Green Version]
- Harvey, A.J.; Crompton, M.R. Use of rna interference to validate brk as a novel therapeutic target in breast cancer: Brk promotes breast carcinoma cell proliferation. Oncogene 2003, 22, 5006–5010. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Shen, C.H.; Tsai, Y.T.; Lin, F.C.; Huang, Y.P.; Chen, R.H. Brk activates rac1 and promotes cell migration and invasion by phosphorylating paxillin. Mol. Cell. Biol. 2004, 24, 10558–10572. [Google Scholar] [CrossRef] [Green Version]
- Lukong, K.E.; Richard, S. Breast tumor kinase brk requires kinesin-2 subunit kap3a in modulation of cell migration. Cell Signal. 2008, 20, 432–442. [Google Scholar] [CrossRef]
- Castro, N.E.; Lange, C.A. Breast tumor kinase and extracellular signal-regulated kinase 5 mediate met receptor signaling to cell migration in breast cancer cells. Breast Cancer Res. 2010, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Irie, H.Y.; Shrestha, Y.; Selfors, L.M.; Frye, F.; Iida, N.; Wang, Z.; Zou, L.; Yao, J.; Lu, Y.; Epstein, C.B.; et al. Ptk6 regulates igf-1-induced anchorage-independent survival. PLoS ONE 2010, 5, e11729. [Google Scholar] [CrossRef]
- Aubele, M.; Walch, A.K.; Ludyga, N.; Braselmann, H.; Atkinson, M.J.; Luber, B.; Auer, G.; Tapio, S.; Cooke, T.; Bartlett, J.M. Prognostic value of protein tyrosine kinase 6 (ptk6) for long-term survival of breast cancer patients. Br. J. Cancer 2008, 99, 1089–1095. [Google Scholar] [CrossRef] [Green Version]
- Aubele, M.; Spears, M.; Ludyga, N.; Braselmann, H.; Feuchtinger, A.; Taylor, K.J.; Lindner, K.; Auer, G.; Stering, K.; Hofler, H.; et al. In situ quantification of her2-protein tyrosine kinase 6 (ptk6) protein-protein complexes in paraffin sections from breast cancer tissues. Br. J. Cancer 2010, 103, 663–667. [Google Scholar] [CrossRef] [Green Version]
- Kamalati, T.; Jolin, H.E.; Mitchell, P.J.; Barker, K.T.; Jackson, L.E.; Dean, C.J.; Page, M.J.; Gusterson, B.A.; Crompton, M.R. Brk, a breast tumor-derived non-receptor protein-tyrosine kinase, sensitizes mammary epithelial cells to epidermal growth factor. J. Biol. Chem. 1996, 271, 30956–30963. [Google Scholar] [CrossRef] [Green Version]
- Kamalati, T.; Jolin, H.E.; Fry, M.J.; Crompton, M.R. Expression of the brk tyrosine kinase in mammary epithelial cells enhances the coupling of egf signalling to pi 3-kinase and akt, via erbb3 phosphorylation. Oncogene 2000, 19, 5471–5476. [Google Scholar] [CrossRef] [Green Version]
- Xiang, B.; Chatti, K.; Qiu, H.; Lakshmi, B.; Krasnitz, A.; Hicks, J.; Yu, M.; Miller, W.T.; Muthuswamy, S.K. Brk is coamplified with erbb2 to promote proliferation in breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 12463–12468. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.H.; Chen, H.Y.; Lin, M.S.; Li, F.Y.; Chang, C.C.; Kuo, M.L.; Settleman, J.; Chen, R.H. Breast tumor kinase phosphorylates p190rhogap to regulate rho and ras and promote breast carcinoma growth, migration, and invasion. Cancer Res. 2008, 68, 7779–7787. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.A.; Lee, E.S.; Yoon, H.Y.; Randazzo, P.A.; Lee, S.T. Ptk6 inhibits down-regulation of egf receptor through phosphorylation of arap1. J. Biol. Chem. 2010, 285, 26013–26021. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, G.; Jain, S.; Kundu, G.C. Osteopontin promotes vascular endothelial growth factor-dependent breast tumor growth and angiogenesis via autocrine and paracrine mechanisms. Cancer Res. 2008, 68, 152–161. [Google Scholar] [CrossRef] [Green Version]
- Miah, S.; Martin, A.; Lukong, K.E. Constitutive activation of breast tumor kinase accelerates cell migration and tumor growth in vivo. Oncogenesis 2012, 1, e11. [Google Scholar] [CrossRef] [Green Version]
- Mercatante, D.R.; Mohler, J.L.; Kole, R. Cellular response to an antisense-mediated shift of bcl-x pre-mrna splicing and antineoplastic agents. J. Biol. Chem. 2002, 277, 49374–49382. [Google Scholar] [CrossRef] [Green Version]
- Ajabnoor, G.M.; Crook, T.; Coley, H.M. Paclitaxel resistance is associated with switch from apoptotic to autophagic cell death in mcf-7 breast cancer cells. Cell Death Dis. 2012, 3, e260. [Google Scholar] [CrossRef] [Green Version]
- Workman, P.; Aboagye, E.O.; Balkwill, F.; Balmain, A.; Bruder, G.; Chaplin, D.J.; Double, J.A.; Everitt, J.; Farningham, D.A.; Glennie, M.J.; et al. Guidelines for the welfare and use of animals in cancer research. Br. J. Cancer 2010, 102, 1555–1577. [Google Scholar] [CrossRef] [Green Version]
- Wazir, U.; Ahmed, M.H.; Bridger, J.M.; Harvey, A.; Jiang, W.G.; Sharma, A.K.; Mokbel, K. The clinicopathological significance of lamin a/c, lamin b1 and lamin b receptor mrna expression in human breast cancer. Cell. Mol. Biol. Lett. 2013, 18, 595–611. [Google Scholar] [CrossRef]
- Pires, I.M.; Blokland, N.J.; Broos, A.W.; Poujade, F.A.; Senra, J.M.; Eccles, S.A.; Span, P.N.; Harvey, A.J.; Hammond, E.M. Hif-1alpha-independent hypoxia-induced rapid ptk6 stabilization is associated with increased motility and invasion. Cancer Biol. Ther. 2014, 15, 1350–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, A.J.; Crompton, M.R. The brk protein tyrosine kinase as a therapeutic target in cancer: Opportunities and challenges. Anticancer Drugs 2004, 15, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, K.A.; Krug, M.; Wersig, T.; Slynko, I.; Schachtele, C.; Totzke, F.; Sippl, W.; Hilgeroth, A. Discovery of 4-anilino alpha-carbolines as novel brk inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 1948–1951. [Google Scholar] [CrossRef] [PubMed]
- Oelze, M.; Mahmoud, K.A.; Sippl, W.; Wersig, T.; Hilgeroth, A.; Ritter, C.A. Novel 4-anilino-alpha-carboline derivatives induce cell death in nonadhesive breast cancer cells through inhibition of brk activity. Int. J. Clin. Pharmacol. Ther. 2015, 53, 1052–1055. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.J.; Barker, K.T.; Shipley, J.; Crompton, M.R. Characterisation and chromosome mapping of the human non receptor tyrosine kinase gene, brk. Oncogene 1997, 15, 1497–1502. [Google Scholar] [CrossRef] [Green Version]
- Danielson, K.G.; Oborn, C.J.; Durban, E.M.; Butel, J.S.; Medina, D. Epithelial mouse mammary cell line exhibiting normal morphogenesis in vivo and functional differentiation in vitro. Proc. Natl. Acad. Sci. USA 1984, 81, 3756–3760. [Google Scholar] [CrossRef] [Green Version]
- Harvey, A.; Burmi, R. Future therapeutic strategies: Implications for brk targeting. In Breast Cancer Current and Alternative Therapeutic Modalities; Gunduz, E., Gunduz, M., Eds.; InTech: London, UK, 2011; pp. 413–434. [Google Scholar]
- Wang, S.; Li, W.; Lv, S.; Wang, Y.; Liu, Z.; Zhang, J.; Liu, T.; Niu, Y. Abnormal expression of nek2 and beta-catenin in breast carcinoma: Clinicopathological correlations. Histopathology 2011, 59, 631–642. [Google Scholar] [CrossRef]
- Palka-Hamblin, H.L.; Gierut, J.J.; Bie, W.; Brauer, P.M.; Zheng, Y.; Asara, J.M.; Tyner, A.L. Identification of beta-catenin as a target of the intracellular tyrosine kinase ptk6. J. Cell Sci. 2010, 123, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Ie Kim, H.; Lee, S.T. Oncogenic functions of ptk6 are enhanced by its targeting to plasma membrane but abolished by its targeting to nucleus. J. Biochem. 2009, 146, 133–139. [Google Scholar] [CrossRef]
- Jiang, J.; Gui, F.; He, Z.; Li, L.; Li, Y.; Li, S.; Wu, X.; Deng, Z.; Sun, X.; Huang, X.; et al. Targeting brk-positive breast cancers with small-molecule kinase inhibitors. Cancer Res. 2017, 77, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Schiff, P.B.; Horwitz, S.B. Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. USA 1980, 77, 1561–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauer, P.M.; Zheng, Y.; Evans, M.D.; Dominguez-Brauer, C.; Peehl, D.M.; Tyner, A.L. The alternative splice variant of protein tyrosine kinase 6 negatively regulates growth and enhances ptk6-mediated inhibition of beta-catenin. PLoS ONE 2011, 6, e14789. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′-’3) | Reverse Primer (5′-’3) |
|---|---|---|
| PTK6V1 | gttctttggctgcatctc | actgaacctgaccgtacattctcgctgaccctgat |
| PTK6V2 | caactacctggccgaga | actgaacctgaccgtacagccctgtggtagttcaca |
| HR Status | Transcript Level (Mean ± SD) | p Value a | ||
|---|---|---|---|---|
| ALT-PTK6 | ER | ER (−) | 320.5 ± 107.0 | 0.15 |
| ER (+) | 181.9 ± 68.9 | |||
| PTK6 | ER (−) | 4.6 ± 2.4 | 0.262 | |
| ER (+) | 3.0 ± 1.6 | |||
| PTK6/ALT-PTK6 ratio | ER (−) | 10,842.3 ± 5207.3 | 0.042 | |
| ER (+) | 4.5 ± 4.5 | |||
| ALT-PTK6 | HER2 | HER2 (−) | 274.1 ± 139.8 | 0.976 |
| HER2 (+) | 266.4 ± 78.4 | |||
| PTK6 | HER2 (−) | 3.7 ± 1.8 | 0.796 | |
| HER2 (+) | 4.0 ± 1.9 | |||
| PTK6/ALT-PTK6 ratio | HER2 (−) | 10,885.8 ± 7001.6 | 0.616 | |
| HER2 (+) | 8937.8 ± 4141.9 | |||
| ALT-PTK6 | PR | PR (−) | 306.1 ± 90.6 | 0.2 |
| PR (+) | 168.1 ± 75.5 | |||
| PTK6 | PR (−) | 4.4 ± 2.0 | 0.314 | |
| PR (+) | 1.4 ± 0.4 | |||
| PTK6/ALT-PTK6 ratio | PR (−) | 9437.09 ± 4479.549 | 0.565 | |
| PR (+) | 6507.15 ± 4462.946 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burmi, R.S.; Box, G.M.; Wazir, U.; Hussain, H.A.; Davies, J.A.; Court, W.J.; Eccles, S.A.; Jiang, W.G.; Mokbel, K.; Harvey, A.J. Breast Tumour Kinase (Brk/PTK6) Contributes to Breast Tumour Xenograft Growth and Modulates Chemotherapeutic Responses In Vitro. Genes 2022, 13, 402. https://doi.org/10.3390/genes13030402
Burmi RS, Box GM, Wazir U, Hussain HA, Davies JA, Court WJ, Eccles SA, Jiang WG, Mokbel K, Harvey AJ. Breast Tumour Kinase (Brk/PTK6) Contributes to Breast Tumour Xenograft Growth and Modulates Chemotherapeutic Responses In Vitro. Genes. 2022; 13(3):402. https://doi.org/10.3390/genes13030402
Chicago/Turabian StyleBurmi, Rajpal S., Gary M. Box, Umar Wazir, Haroon A. Hussain, Julie A. Davies, William J. Court, Suzanne A. Eccles, Wen G. Jiang, Kefah Mokbel, and Amanda J. Harvey. 2022. "Breast Tumour Kinase (Brk/PTK6) Contributes to Breast Tumour Xenograft Growth and Modulates Chemotherapeutic Responses In Vitro" Genes 13, no. 3: 402. https://doi.org/10.3390/genes13030402