
Maternal Uniparental Isodisomy of Chromosome 4 and 8 in Patients with Retinal Dystrophy: SRD5A3-Congenital Disorders of Glycosylation and RP1-Related Retinitis Pigmentosa

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Genetic Analysis
2.3. Electrospray Ionization Mass Spectrometry
2.4. Clinical Assessment
3. Results
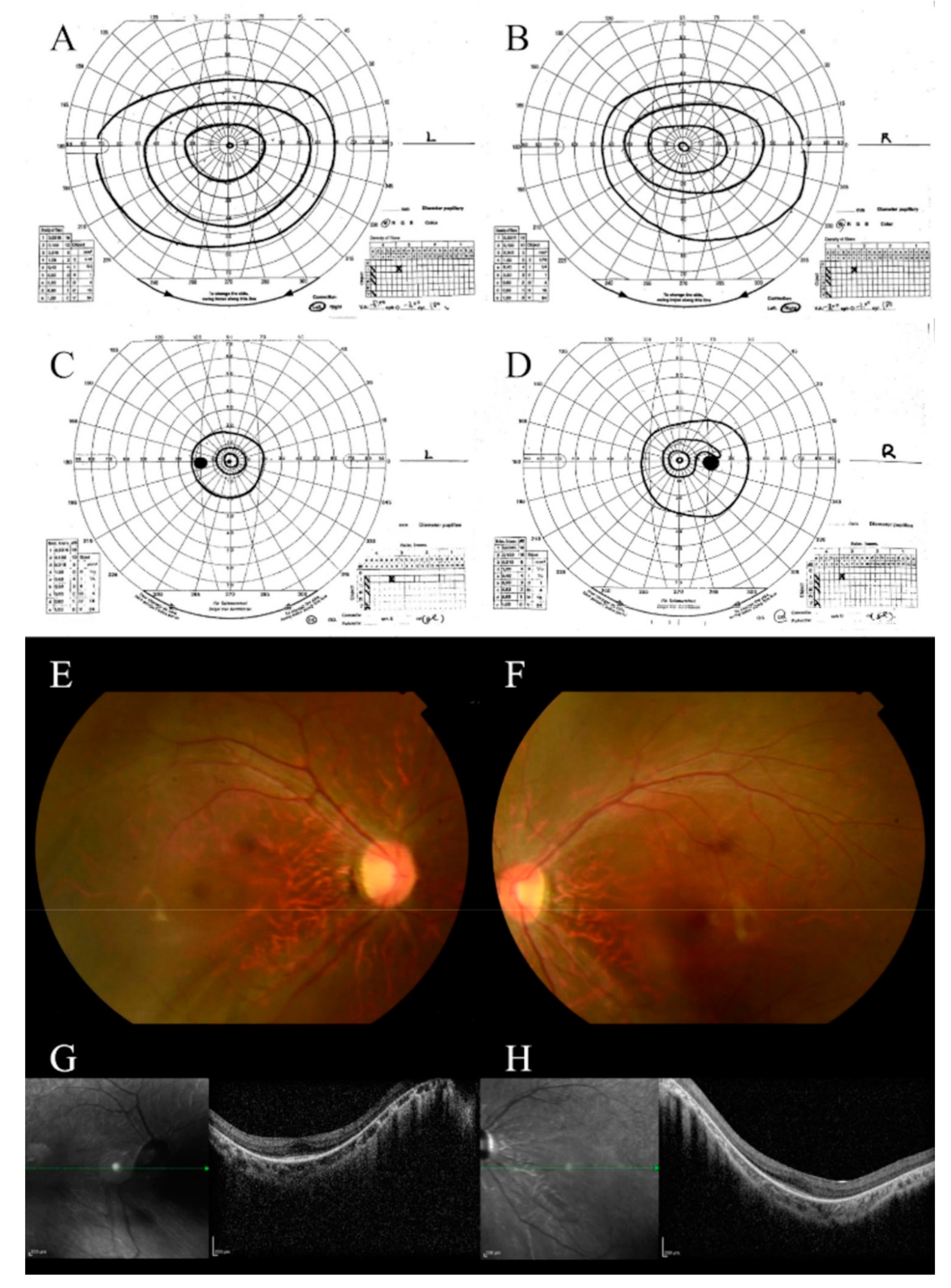
3.1. Case 1
3.1.1. Clinical Characteristics
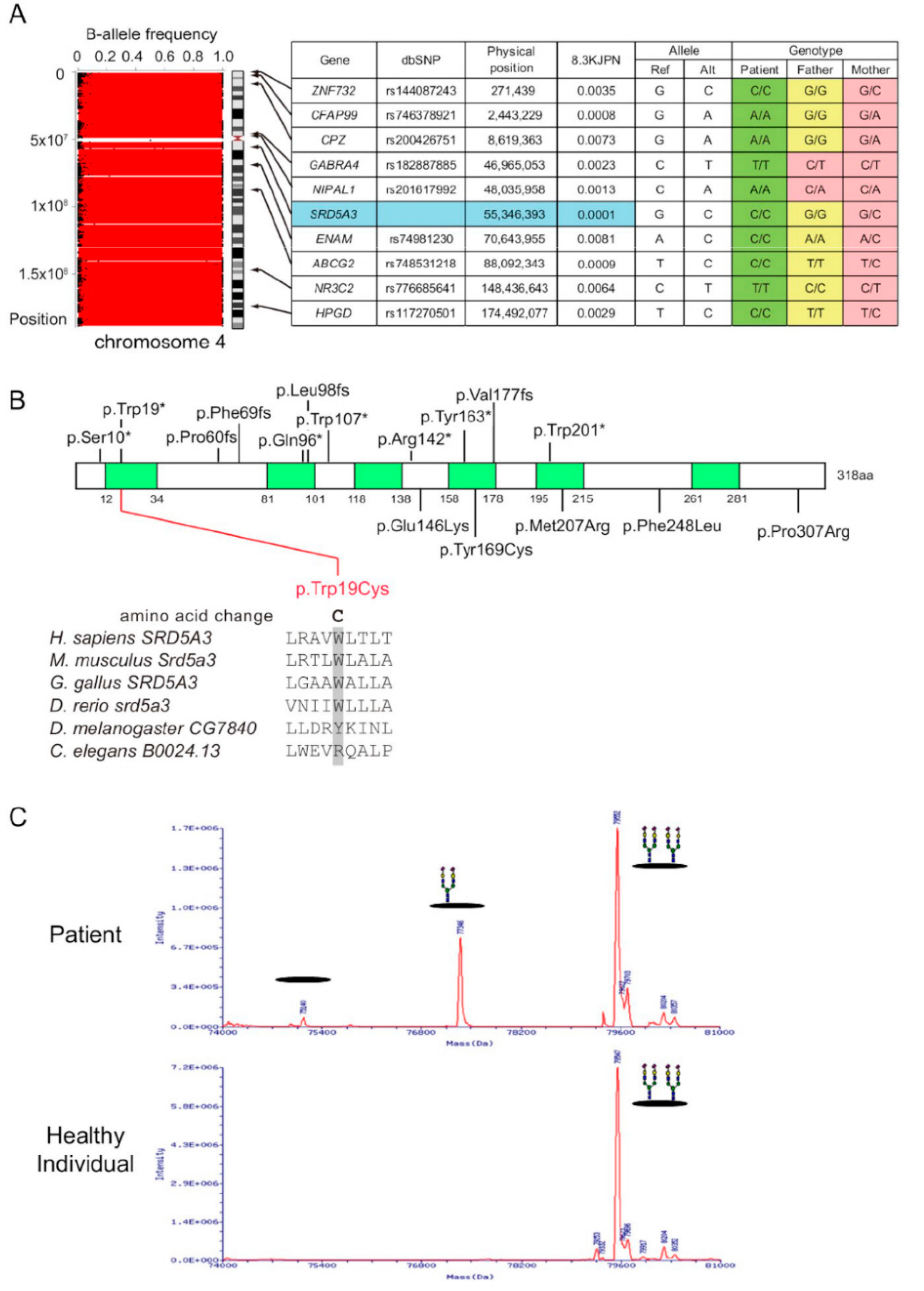
3.1.2. Genetic Studies
3.1.3. Mass Spectrum of Transferrin
3.2. Case 2
3.2.1. Clinical Characteristics
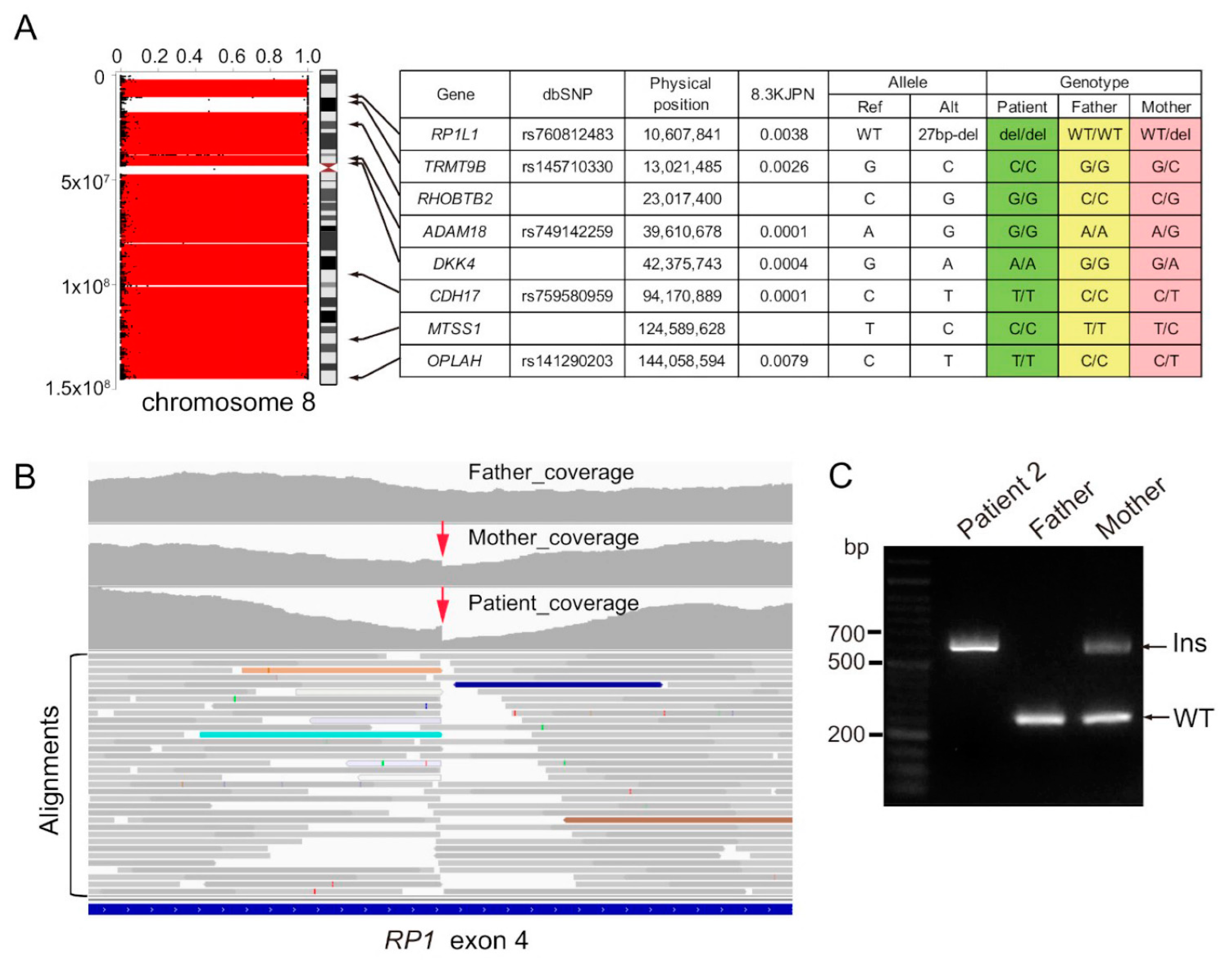
3.2.2. Genetic Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Engel, E. A New Genetic Concept: Uniparental Disomy and Its Potential Effect, Isodisomy. Am. J. Med. Genet. 1980, 6, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Liehr, T. Cytogenetic Contribution to Uniparental Disomy (UPD). Mol. Cytogenet. 2010, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, J.E.; Perciaccante, R.G.; Greig, G.M.; Willard, H.F.; Ledbetter, D.H.; Hejtmancik, J.F.; Pollack, M.S.; O’Brien, W.E.; Beaudet, A.L. Uniparental Disomy as a Mechanism for Human Genetic Disease. Am. J. Hum. Genet. 1988, 42, 217–226. [Google Scholar] [PubMed]
- Eggermann, T. Prenatal Detection of Uniparental Disomies (UPD): Intended and Incidental Finding in the Era of Next Generation Genomics. Genes 2020, 11, 1454. [Google Scholar] [CrossRef]
- Mutirangura, A.; Greenberg, F.; Butler, M.G.; Malcolm, S.; Nicholls, R.D.; Chakravarti, A.; Ledbetter, D.H. Multiplex PCR of Three Dinucleotide Repeats in the Prader-Willi/Angelman Critical Region (15q11-Q13): Molecular Diagnosis and Mechanism of Uniparental Disomy. Hum. Mol. Genet. 1993, 2, 143–151. [Google Scholar] [CrossRef]
- Yamazawa, K.; Ogata, T.; Ferguson-Smith, A.C. Uniparental Disomy and Human Disease: An Overview. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 329–334. [Google Scholar] [CrossRef]
- Ogata, T.; Kagami, M. Molecular Mechanisms Leading to the Phenotypic Development in Paternal and Maternal Uniparental Disomy for Chromosome 14. Clin. Pediatr. Endocrinol. 2008, 17, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Engel, E. A Fascination with Chromosome Rescue in Uniparental Disomy: Mendelian Recessive Outlaws and Imprinting Copyrights Infringements. Eur. J. Hum. Genet. 2006, 14, 1158–1169. [Google Scholar] [CrossRef] [Green Version]
- Pentao, L.; Lewis, R.A.; Ledbetter, D.H.; Patel, P.I.; Lupski, J.R. Maternal Uniparental Isodisomy of Chromosome 14: Association with Autosomal Recessive Rod Monochromacy. Am. J. Hum. Genet. 1992, 50, 690–699. [Google Scholar]
- Thompson, D.A.; McHenry, C.L.; Li, Y.; Richards, J.E.; Othman, M.I.; Schwinger, E.; Vollrath, D.; Jacobson, S.G.; Gal, A. Retinal Dystrophy Due to Paternal Isodisomy for Chromosome 1 or Chromosome 2, with Homoallelism for Mutations in RPE65 or MERTK, Respectively. Am. J. Hum. Genet. 2002, 70, 224–229. [Google Scholar] [CrossRef] [Green Version]
- Rivolta, C.; Berson, E.L.; Dryja, T.P. Paternal Uniparental Heterodisomy with Partial Isodisomy of Chromosome 1 in a Patient with Retinitis Pigmentosa Without Hearing Loss and a Missense Mutation in the Usher Syndrome type II Gene USH2A. Arch. Ophthalmol. 2002, 120, 1566–1571. [Google Scholar] [CrossRef] [Green Version]
- Fingert, J.H.; Eliason, D.A.; Phillips, N.C.; Lotery, A.J.; Sheffield, V.C.; Stone, E.M. Case of Stargardt Disease Caused by Uniparental Isodisomy. Arch. Ophthalmol. 2006, 124, 744–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiszniewski, W.; Lewis, R.A.; Lupski, J.R. Achromatopsia: The CNGB3 p.T383fsX Mutation Results from a Founder Effect and Is Responsible for the Visual Phenotype in the Original Report of Uniparental Disomy 14. Hum. Genet. 2007, 121, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Riveiro-Alvarez, R.; Valverde, D.; Lorda-Sanchez, I.; Trujillo-Tiebas, M.J.; Cantalapiedra, D.; Vallespin, E.; Aguirre-Lamban, J.; Ramos, C.; Ayuso, C. Partial Paternal Uniparental Disomy (UPD) of Chromosome 1 in a Patient with Stargardt Disease. Mol. Vis. 2007, 13, 96–101. [Google Scholar] [PubMed]
- Roosing, S.; van den Born, L.I.; Hoyng, C.B.; Thiadens, A.A.; de Baere, E.; Collin, R.W.; Koenekoop, R.K.; Leroy, B.P.; van Moll-Ramirez, N.; Venselaar, H.; et al. Maternal Uniparental Isodisomy of Chromosome 6 Reveals a TULP1 Mutation as a Novel Cause of Cone Dysfunction. Ophthalmology. 2013, 120, 1239–1246. [Google Scholar] [CrossRef]
- Souzeau, E.; Thompson, J.A.; McLaren, T.L.; De Roach, J.N.; Barnett, C.P.; Lamey, T.M.; Craig, J.E. Maternal Uniparental Isodisomy of Chromosome 6 Unmasks a Novel Variant in TULP1 in a Patient with Early Onset Retinal Dystrophy. Mol. Vis. 2018, 24, 478–484. [Google Scholar]
- Kohl, S.; Baumann, B.; Dassie, F.; Mayer, A.K.; Solaki, M.; Reuter, P.; Kühlewein, L.; Wissinger, B.; Maffei, P. Paternal Uniparental Isodisomy of Chromosome 2 in a Patient with CNGA3-Associated Autosomal Recessive Achromatopsia. Int. J. Mol. Sci. 2021, 22, 7842. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Tadaka, S.; Hishinuma, E.; Komaki, S.; Motoike, I.N.; Kawashima, J.; Saigusa, D.; Inoue, J.; Takayama, J.; Okamura, Y.; Aoki, Y.; et al. jMorp Updates in 2020: Large Enhancement of Multi-Omics Data Resources on the General Japanese Population. Nucleic Acids Res. 2021, 49, D536–D544. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magi, A.; Tattini, L.; Palombo, F.; Benelli, M.; Gialluisi, A.; Giusti, B.; Abbate, R.; Seri, M.; Gensini, G.F.; Romeo, G.; et al. H3M2: Detection of Runs of Homozygosity from Whole-Exome Sequencing Data. Bioinformatics 2014, 30, 2852–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masunaga, Y.; Mochizuki, M.; Kadoya, M.; Wada, Y.; Okamoto, N.; Fukami, M.; Kato, F.; Saitsu, H.; Ogata, T. Primary Ovarian Insufficiency in a Female with phosphomannomutase-2 Gene (PMM2) Mutations for Congenital Disorder of Glycosylation. Endocr. J. 2021, 68, 605–611. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for Full-Field Clinical Electroretinography (2015 Update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cantagrel, V.; Lefeber, D.J.; Ng, B.G.; Guan, Z.; Silhavy, J.L.; Bielas, S.L.; Lehle, L.; Hombauer, H.; Adamowicz, M.; Swiezewska, E.; et al. SRD5A3 Is Required for Converting Polyprenol to Dolichol and Is Mutated in a Congenital Glycosylation Disorder. Cell 2010, 142, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Nikopoulos, K.; Cisarova, K.; Quinodoz, M.; Koskiniemi-Kuendig, H.; Miyake, N.; Farinelli, P.; Rehman, A.U.; Khan, M.I.; Prunotto, A.; Akiyama, M.; et al. A Frequent Variant in the Japanese Population Determines Quasi-Mendelian Inheritance of Rare Retinal Ciliopathy. Nat. Commun. 2019, 10, 2884. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.M.; Stenson, P.D.; Cooper, D.N.; Férec, C. A Systematic Analysis of LINE-1 Endonuclease-Dependent Retrotranspositional Events Causing Human Genetic Disease. Hum. Genet. 2005, 117, 411–427. [Google Scholar] [CrossRef]
- Morava, E.; Wevers, R.A.; Cantagrel, V.; Hoefsloot, L.H.; Al-Gazali, L.; Schoots, J.; van Rooij, A.; Huijben, K.; van Ravenswaaij-Arts, C.M.; Jongmans, M.C.; et al. A Novel Cerebello-Ocular Syndrome with Abnormal Glycosylation Due to Abnormalities in Dolichol Metabolism. Brain 2010, 133, 3210–3220. [Google Scholar] [CrossRef]
- Taylor, R.L.; Arno, G.; Poulter, J.A.; Khan, K.N.; Morarji, J.; Hull, S.; Pontikos, N.; Rueda Martin, A.; Smith, K.R.; Ali, M.; et al. and the 100,000 Genomes Project. Association of Steroid 5α-Reductase type 3 Congenital Disorder of Glycosylation with Early-Onset Retinal Dystrophy. JAMA Ophthalmol. 2017, 135, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.O. Early-Onset Retinal Dystrophy and Chronic Dermatitis in a Girl with an Undiagnosed Congenital Disorder of Glycosylation (SRD5A3-CDG). Ophthalmic Genet. 2018, 39, 628–630. [Google Scholar] [CrossRef]
- Kousal, B.; Honzík, T.; Hansíková, H.; Ondrušková, N.; Čechová, A.; Tesařová, M.; Stránecký, V.; Meliška, M.; Michaelides, M.; Lišková, P. Review of SRD5A3 Disease-Causing Sequence Variants and Ocular Findings in Steroid 5α-Reductase type 3 Congenital Disorder of Glycosylation, and a Detailed New Case. Folia Biol. 2019, 65, 134–141. [Google Scholar]
- Audo, I.; Mohand-Saïd, S.; Dhaenens, C.M.; Germain, A.; Orhan, E.; Antonio, A.; Hamel, C.; Sahel, J.A.; Bhattacharya, S.S.; Zeitz, C. RP1 and Autosomal Dominant Rod-Cone Dystrophy: Novel Mutations, a Review of Published Variants, and Genotype-Phenotype correlation. Hum. Mutat. 2012, 33, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Collin, R.W.; Cremers, F.P.; den Hollander, A.I.; van den Born, L.I.; Pierce, E.A. Expression of Wild-Type Rp1 Protein in Rp1 Knock-In Mice Rescues the Retinal Degeneration Phenotype. PLoS ONE 2012, 7, e43251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemiatkowska, A.M.; Astuti, G.D.; Arimadyo, K.; den Hollander, A.I.; Faradz, S.M.; Cremers, F.P.; Collin, R.W. Identification of a Novel Nonsense Mutation in RP1 That Causes Autosomal Recessive Retinitis Pigmentosa in an Indonesian Family. Mol. Vis. 2012, 18, 2411–2419. [Google Scholar] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; den Hollander, A.I.; Geerlings, M.J.; Kersten, E.; Klevering, B.J.; Klaver, C.C.W.; Plomp, A.S.; Wesseling, N.L.; Bergen, A.A.B.; et al. Macular Dystrophy and Cone-Rod Dystrophy Caused by Mutations in the RP1 Gene: Extending the RP1 Disease Spectrum. Invest. Ophthalmol. Vis. Sci. 2019, 60, 1192–1203. [Google Scholar] [CrossRef] [Green Version]
- Mizobuchi, K.; Hayashi, T.; Oishi, N.; Kubota, D.; Kameya, S.; Higasa, K.; Futami, T.; Kondo, H.; Hosono, K.; Kurata, K.; et al. Genotype-Phenotype Correlations in RP1-Associated Retinal Dystrophies: A Multi-Center Cohort Study in JAPAN. J. Clin. Med. 2021, 10, 2265. [Google Scholar] [CrossRef]
- Kurata, K.; Hosono, K.; Hotta, Y. Clinical and Genetic Findings of a Japanese Patient with RP1-Related Autosomal Recessive Retinitis Pigmentosa. Doc. Ophthalmol. 2018, 137, 47–56. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHR | Sex | Onset | Disomy | Gene | Origin | Ocular Diagnosis | Nystagmus | Ocular Findings | Intellectual Disability | Other Systemic Abnormalities | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | male | <5 years | Whole | RPE65 | paternal | LCA | + | VA: 20/200 in both eyes with a refraction of +4.00 (8 y) ERG: rod and cone responses were not detectable Prognosis: small islands of vision remaining (50 y) | - | hyperbetalipoproteinemiabenign hyperbilirubinemia | [10] |
| 1 | female | - | Partial | USH2A | paternal | RP | - | VA: 20/40 in both eyes with a mean refraction of +1.50 (49 y) VF: V4e were constricted to a central island extending to the 8° ERG: rod-plus-cone and cone ERG amplitudes were remarkably decreased | - | no hearing loss | [11] |
| 1 | female | 15 years | Whole | ABCA4 | paternal | Stargardt disease | - | VA: 20/200 OD and 20/150 OS Fundus: macular and extramacular pisciform yellow flecks VF: bilateral central scotomas ERG: normal | - | - | [12] |
| 1 | female | 2–3 years | Partial | ABCA4 | paternal | Stargardt disease | - | mild vision loss and strabismus (2–3 y), photophobia and dyschromatopsia VA: 100/200 Fundus: yellowish flecks at the macula | - | - | [14] |
| 2 | female | 3–5 years | Whole | MERTK | paternal | RP | - | night blindness, poor vision (preschool), peripheral vision reduction Prognosis: 5° visual field (34 y) | - | - | [10] |
| 2 | female | first week | Partial or Whole | CNGA3 | paternal | ACHM | + | photophobia Color vision tests: findings typical for ACHM ERG: severely reduced cone and responses Prognosis: Visual disturbances remained stable | - | multiple bilateral kidney cysts with a maximum diameter of 14 mm | [17] |
| 4 | female | birth | Whole | SRD5A3 | maternal | RD | + | Fundus: poor retinal color and narrowing of the retinal blood vessels VF: remarkably constricted (14 y) ERG: extinguished pattern | + | epilepsy | this study |
| 6 | male | 43 years | Whole | TULP1 | maternal | RD with cone dysfunction | - | severe color vision defects VA: 80/200 OD and 120/200 OS (at onset) VF: central scotoma Fundus: macular bull’s eye, peripheral mottling vessels ERG: relatively preserved ERG Prognosis: CF OD and 10/200 OS (52 y) | - | intrauterine growth retardation | [15] |
| 6 | female | 3 months | Whole | TULP1 | maternal | RD with rod-cone dysfunction | + | poor pupillary constriction to strong light retinal pallor (3 months) poor night vision and peripheral vision VA: 6/76 in both eyes Fundus: RP-like, no optic disc atrophy ERG: non-recordable (17 months) | - | intrauterine growth retardation | [16] |
| 8 | male | 7 years | Whole | RP1 | maternal | RP | - | photophobia and night blindness (7 y) VF: highly constricted Fundus: typical findings RP, and degenerated lesions in the macula FAF: low fluorescence consistent with retinal degeneration ERG: extinguished pattern | - | - | this study |
| 14 | female | 5 days | Partial | CNGB3 | maternal | ACHM | + | A-pattern exotropia, sluggish pupils without afferent defect progressive compound myopic astigmatic refractive error VA: 20/160 in both eyes Color vision test: findings typical for ACHM ERG: no recordable cone function, normal rod function | + | short stature, minimal dysmorphism premature puberty, small hands and feet reproductive history of three consecutive first-trimester miscarriages | [9,13] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tachibana, N.; Hosono, K.; Nomura, S.; Arai, S.; Torii, K.; Kurata, K.; Sato, M.; Shimakawa, S.; Azuma, N.; Ogata, T.; et al. Maternal Uniparental Isodisomy of Chromosome 4 and 8 in Patients with Retinal Dystrophy: SRD5A3-Congenital Disorders of Glycosylation and RP1-Related Retinitis Pigmentosa. Genes 2022, 13, 359. https://doi.org/10.3390/genes13020359
Tachibana N, Hosono K, Nomura S, Arai S, Torii K, Kurata K, Sato M, Shimakawa S, Azuma N, Ogata T, et al. Maternal Uniparental Isodisomy of Chromosome 4 and 8 in Patients with Retinal Dystrophy: SRD5A3-Congenital Disorders of Glycosylation and RP1-Related Retinitis Pigmentosa. Genes. 2022; 13(2):359. https://doi.org/10.3390/genes13020359
Chicago/Turabian StyleTachibana, Nobutaka, Katsuhiro Hosono, Shuhei Nomura, Shinji Arai, Kaoruko Torii, Kentaro Kurata, Miho Sato, Shuichi Shimakawa, Noriyuki Azuma, Tsutomu Ogata, and et al. 2022. "Maternal Uniparental Isodisomy of Chromosome 4 and 8 in Patients with Retinal Dystrophy: SRD5A3-Congenital Disorders of Glycosylation and RP1-Related Retinitis Pigmentosa" Genes 13, no. 2: 359. https://doi.org/10.3390/genes13020359