
Altered Mitochondrial Quality Control in Rats with Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD) Induced by High-Fat Feeding

 , ,
, , 


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
- The first group, control group (N), received a standard diet ad libitum (total metabolizable percentage of energy: 60.4 carbohydrates, 29 proteins, and 10.6 fat J−1; 15.88 kJ gross energy g−1) (Muscedola, Milan, Italy) for fourteen weeks;
- The second group, MAFLD group (HFD) received a high-fat diet (HFD) ad libitum (280 g diet supplemented with 395 g of lyophilized lamb meat (Liomellin, Milan, Italy), 120 g cellulose (Sigma-Aldrich, St. Louis, MO, USA), 20 g mineral mix (ICN Biomedical, Solon, OH, USA), 7 g vitamin mix (ICN), and 200 g low-salt butter (Lurpak, Denmark); total metabolizable percentage of energy: 21 carbohydrates, 29 proteins, and 50 fat J−1; 19.85 kJ gross energy g−1) for fourteen weeks to reproduce a model of overweight recently characterized for visceral adipose tissue inflammation [27].
2.2. Indirect Calorimetry
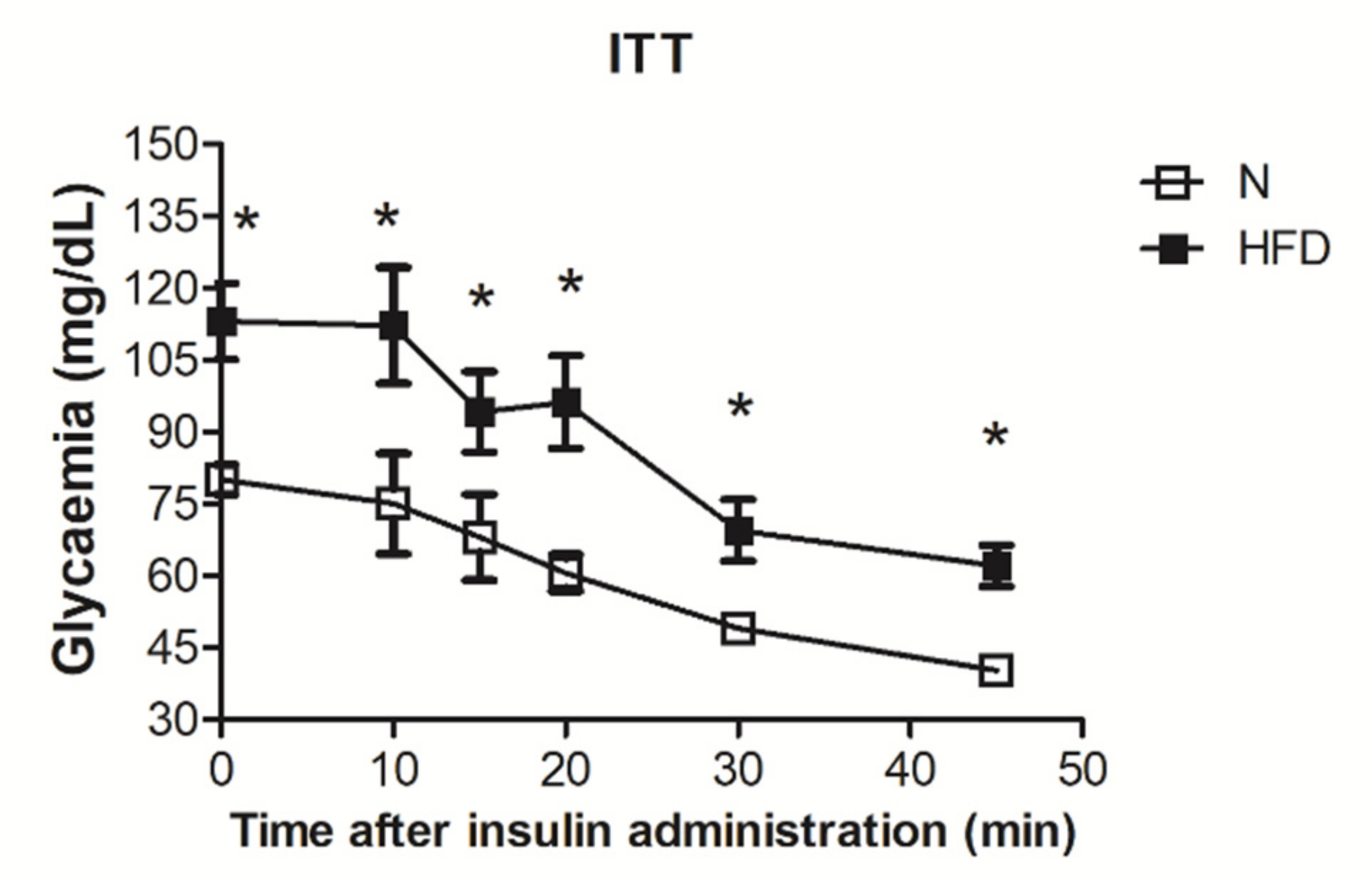
2.3. Insulin Tolerance Test
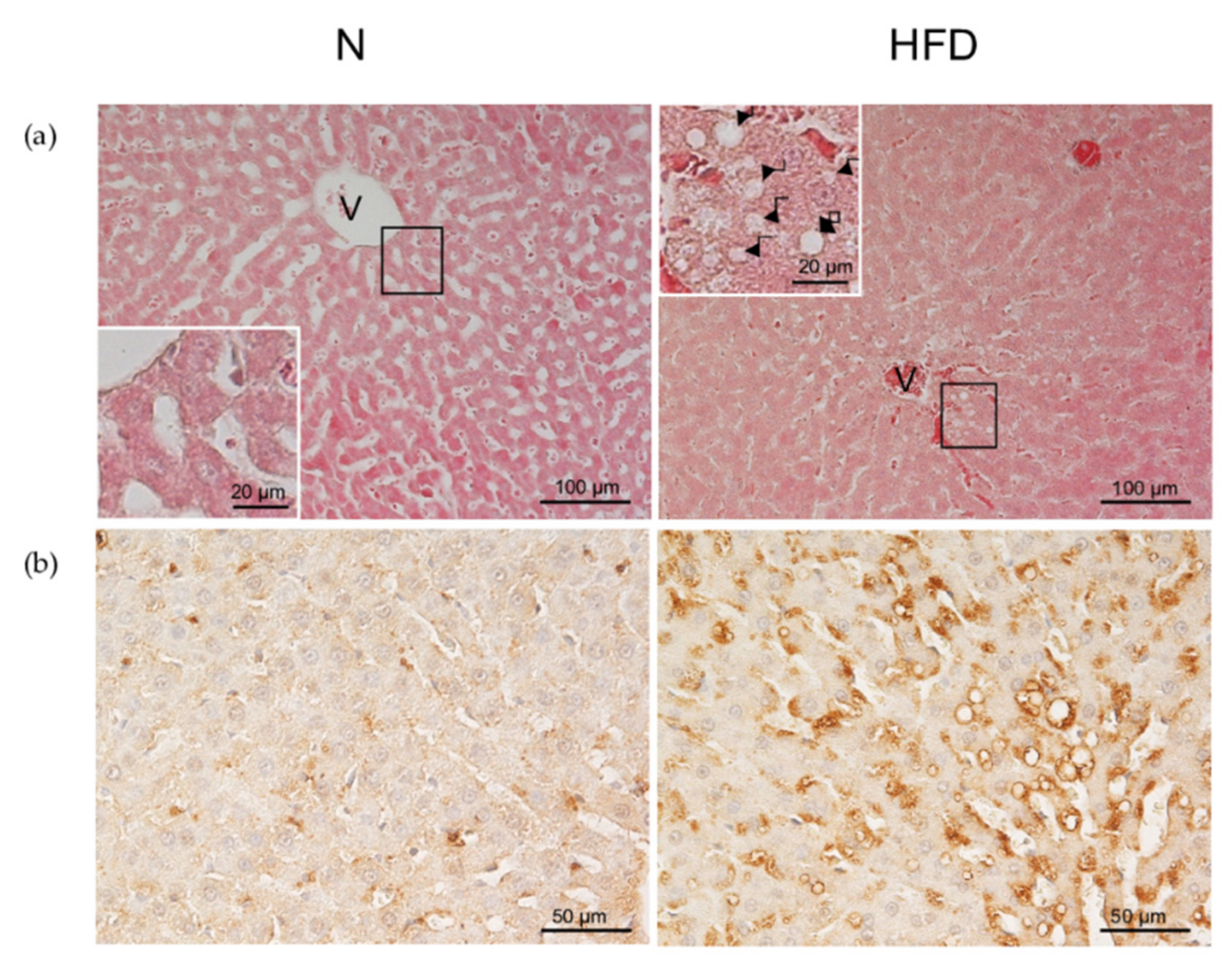
2.4. Liver Hystology
2.5. Immunohistochemical Staining for ADRP
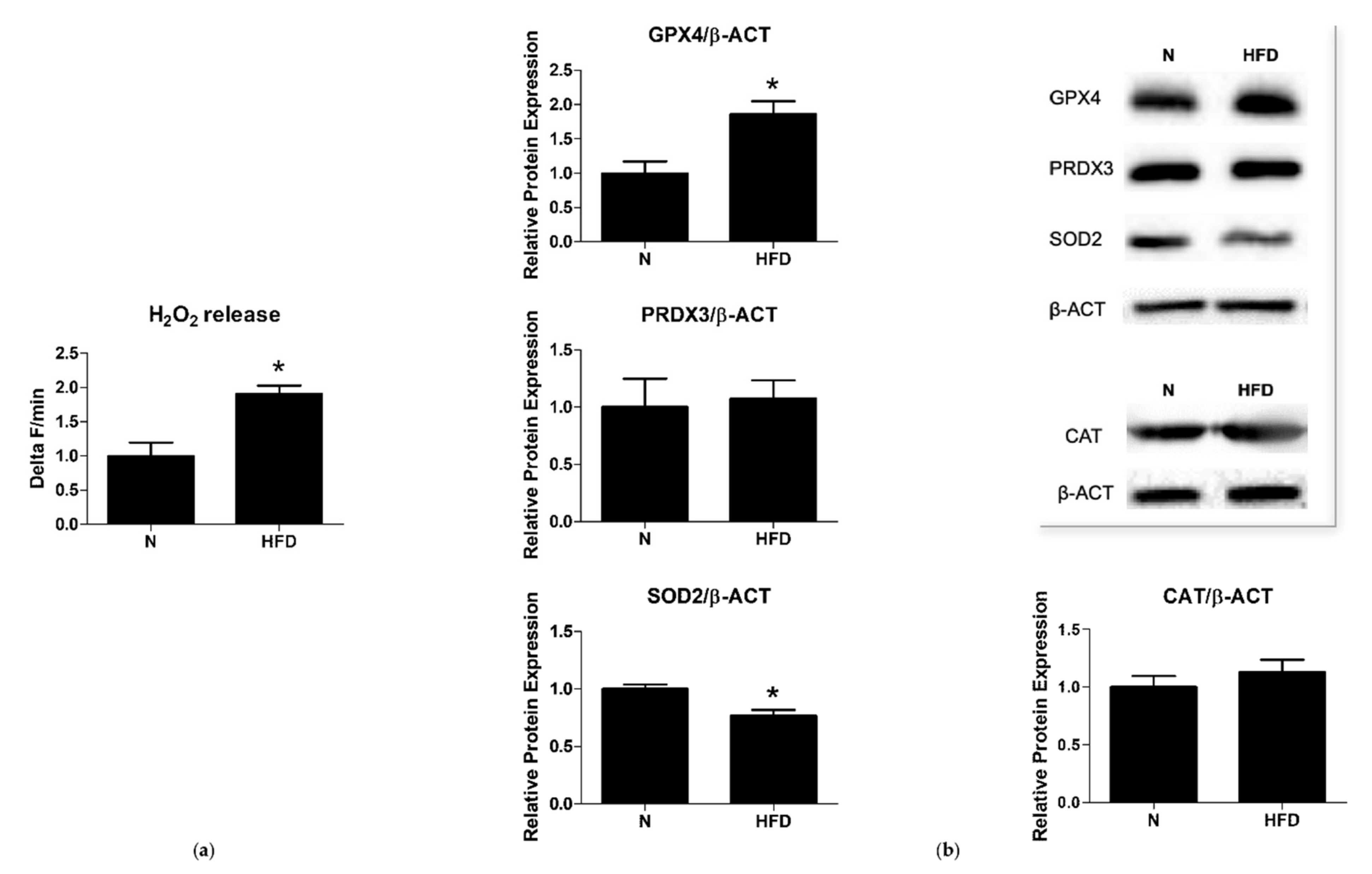
2.6. Mitochondrial H2O2 Release
2.7. Genomic DNA Isolation
2.8. Quantitative Polymerase Chain Reaction (QPCR)
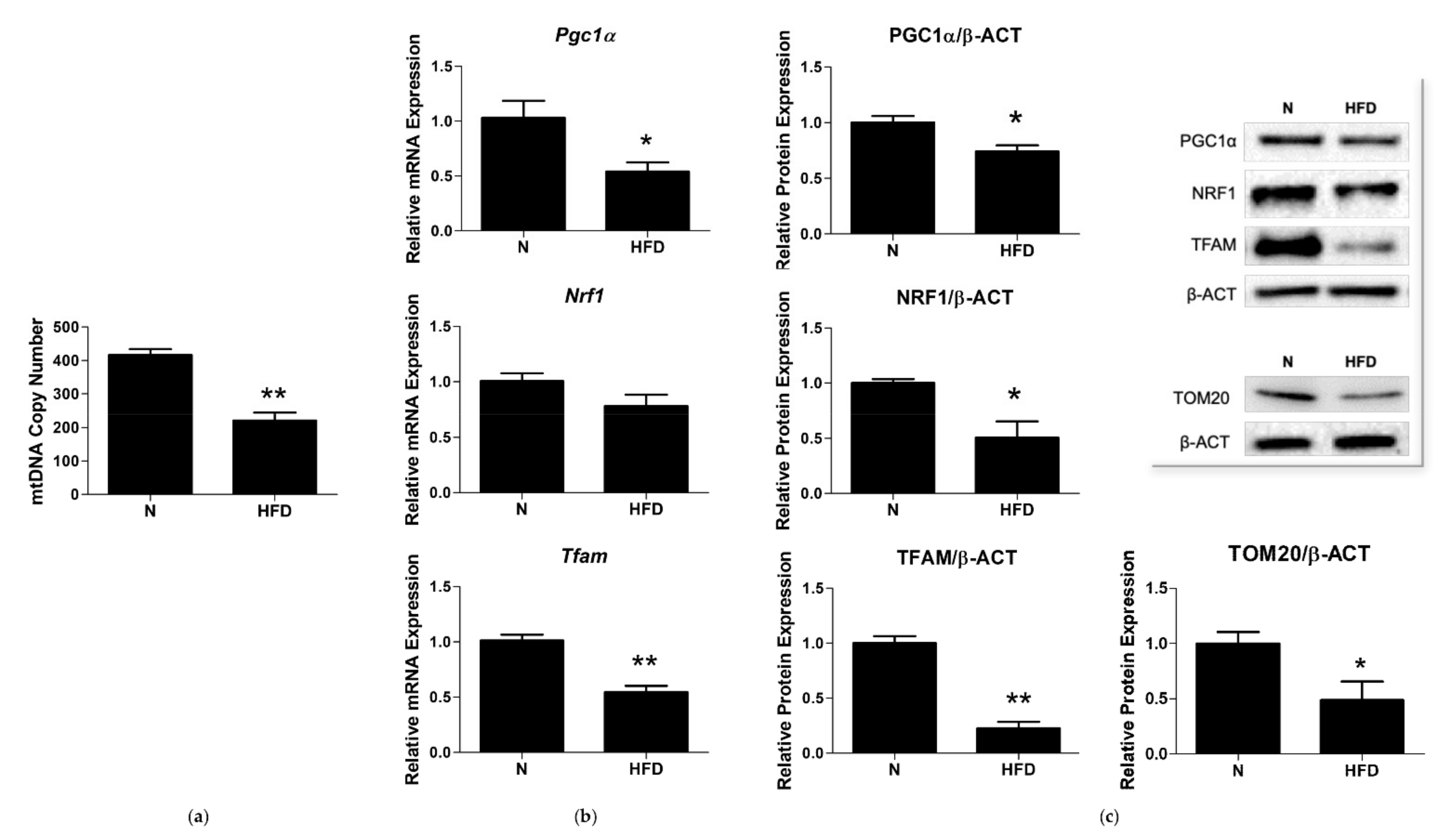
2.9. Quantification of mtDNA Copy Number by Real-Time PCR
2.10. Total RNA Isolation from Liver and qRT-PCR
2.11. Western Blot Analysis
2.12. Statistical Analysis
3. Results
3.1. Establishment of the MAFDL Rat Model
3.2. Mitochondrial ROS Production and Antioxidant Enzymes
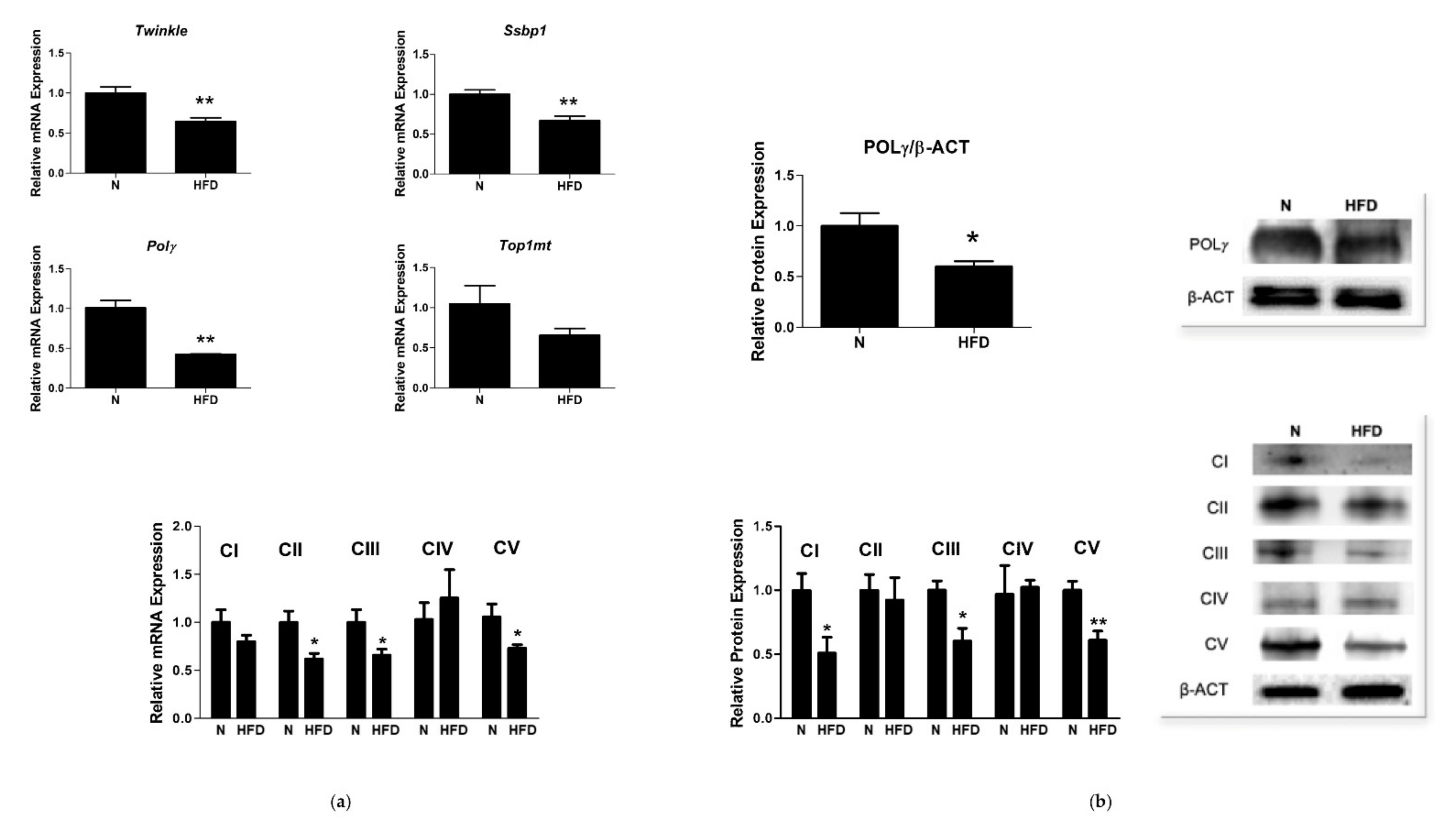
3.3. mtDNA Copy Number and Expression of Markers involved in Mitochondrial Biogenesis and Replisome
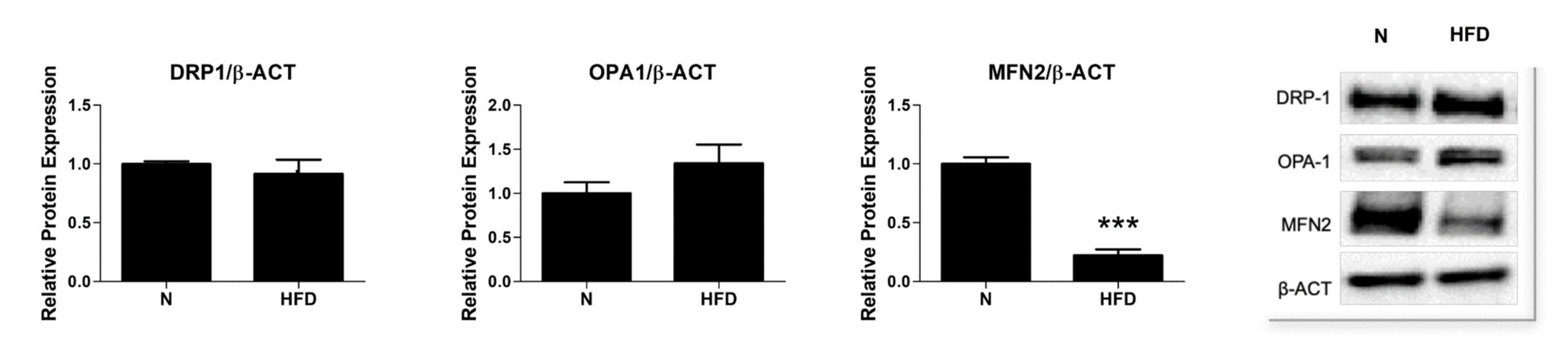
3.4. Expression of Proteins Involved in Mitochondrial Dynamics
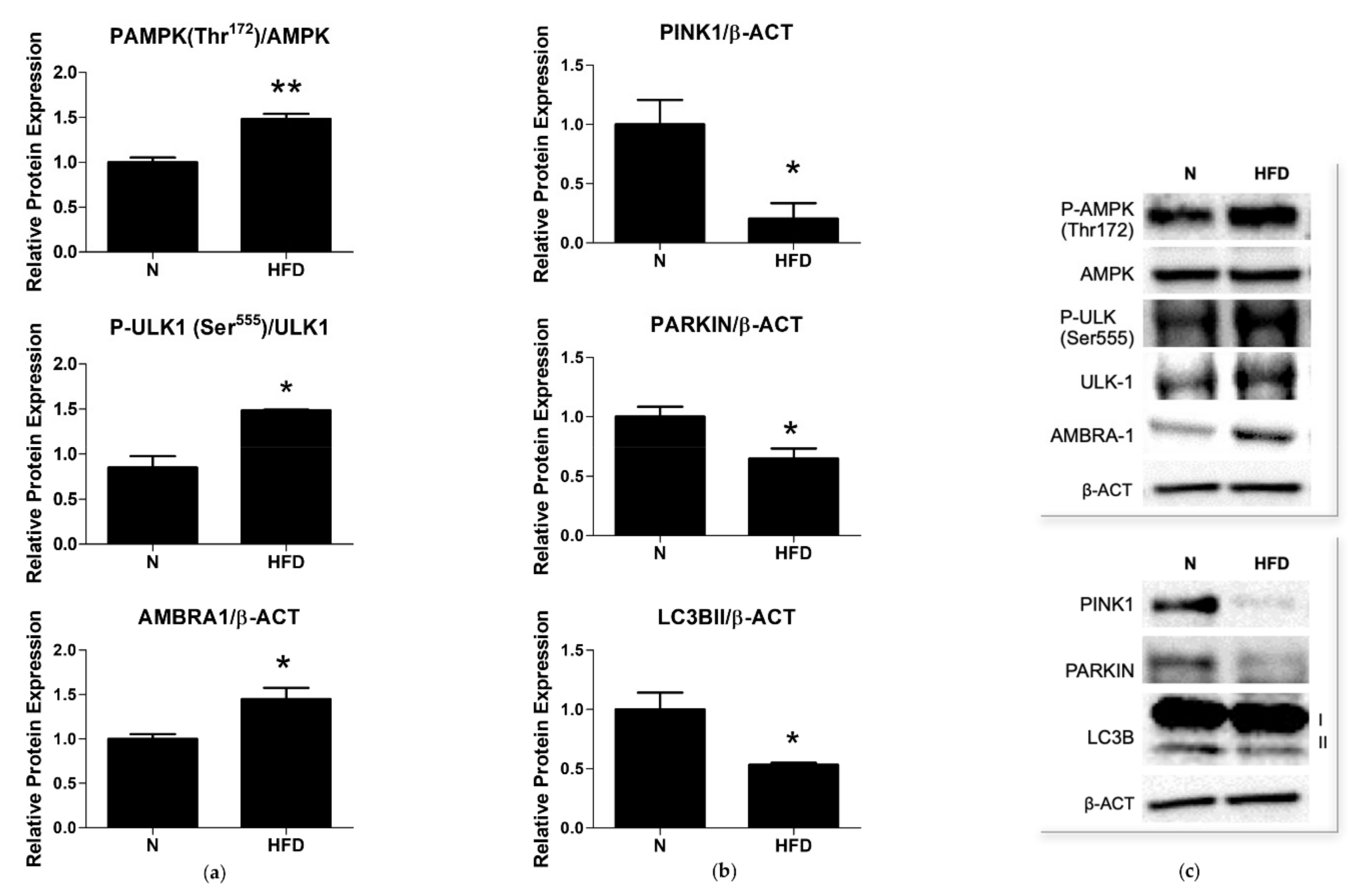
3.5. Expression of Markers of Mitophagy
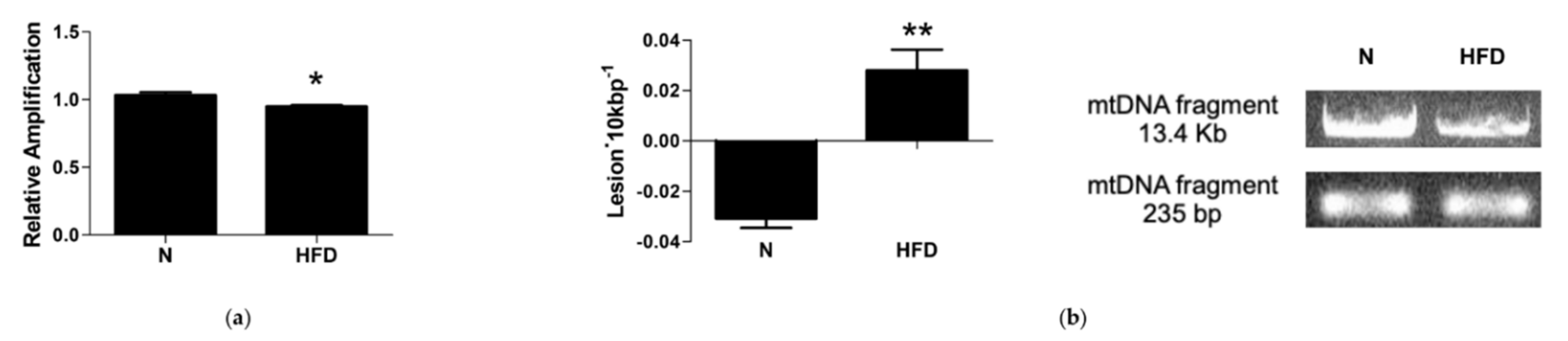
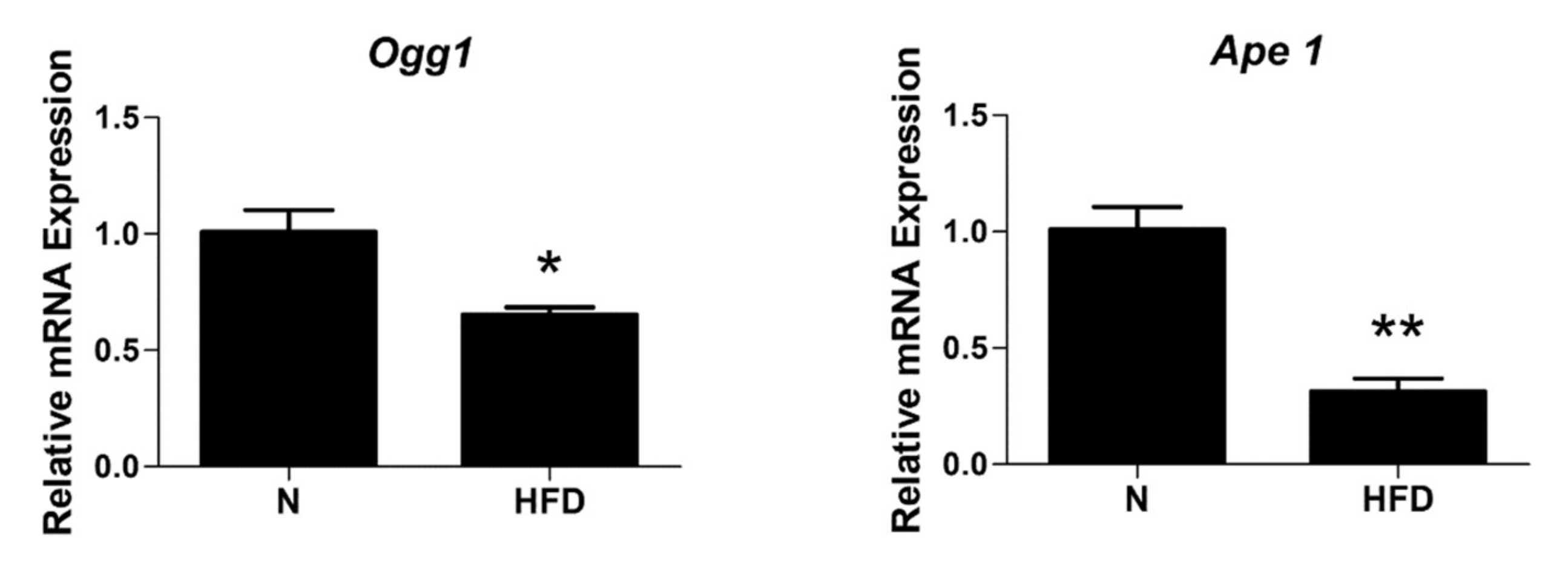
3.6. mtDNA Damage and Lesion Frequency
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Sarin, S.K.; Kumar, M.; Eslam, M.; George, J.; Al Mahtab, M.; Akbar, S.M.F.; Jia, J.; Tian, Q.; Aggarwal, R.; Muljono, D.; et al. Liver diseases in the Asia-Pacific region: A Lancet Gastroenterology & hepatology Commission. Lancet Gastroenterol. Hepatol. 2020, 5, 167–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recena Aydos, L.; Aparecida do Amaral, L.; Serafim de Souza, R.; Jacobowski, A.C.; Freitas Dos Santos, E.; Rodrigues Macedo, M.L. Nonalcoholic Fatty Liver Disease Induced by High-Fat Diet in C57bl/6 Models. Nutrients 2019, 11, 3067. [Google Scholar] [CrossRef] [Green Version]
- White, P.A.S.; Cercato, L.M.; Araújo, J.M.D.; Souza, L.A.; Soares, A.F.; Barbosa, A.P.O.; Neto, J.M.D.R.; Marçal, A.C.; Machado, U.F.; Camargo, E.A.; et al. Modelo de obesidade induzida por dieta hiperlipídica e associada à resistência à ação da insulina e intolerância à glicose. Arq. Bras. Endocrinol. Metabol. 2013, 57, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Rocchini, A.P.; Marker, P.; Cervenka, T. Time course of insulin resistance associated with feeding dogs a high- fat diet. Am. J. Physiol. Endocrinol. Metab. 1997, 272, E147–E154. [Google Scholar] [CrossRef]
- Buettner, R.; Schölmerich, J.; Bollheimer, L.C. High-fat diets: Modeling the metabolic disorders of human obesity in rodents. Obesity 2007, 15, 798–808. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of Insulin Resistance in MAFLD. Int. J. Mol. Sci. 2021, 22, 4156. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Zhang, H.X.; Guo, J.R.; Lam, C.W.K.; Wang, C.Y.; Zhang, W. Mitochondria-Mediated Pathogenesis and Therapeutics for Non-Alcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, e1900043. [Google Scholar] [CrossRef]
- Bruce, K.D.; Cagampang, F.R.; Argenton, M.; Zhang, J.; Ethirajan, P.L.; Burdge, G.C.; Bateman, A.C.; Clough, G.F.; Poston, L.; Hanson, M.A.; et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009, 50, 1796–1808. [Google Scholar] [CrossRef]
- Morio, B.; Panthu, B.; Bassot, A.; Rieusset, J. Role of mitochondria in liver metabolic health and diseases. Cell Calcium. 2021, 94, 102336. [Google Scholar] [CrossRef]
- Grattagliano, I.; Montezinho, L.P.; Oliveira, P.J.; Frühbeck, G.; Gómez-Ambrosi, J.; Montecucco, F.; Carbone, F.; Wieckowski, M.R.; Wang, D.Q.; Portincasa, P. Targeting mitochondria to oppose the progression of nonalcoholic fatty liver disease. Biochem. Pharmacol. 2019, 160, 34–45. [Google Scholar] [CrossRef]
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, hypertension, and cardiovascular disease: Clinical insights and vascular mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Grundy, S.M. Atherogenic dyslipidemia associated with metabolic syndrome and insulin resistance. Clin. Cornerstone 2006, 8 (Suppl. 1), S21–S27. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Druzhyna, N.M.; Wilson, G.L.; LeDoux, S.P. Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 2008, 129, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Flichman, D.; Scian, R.; Rohr, C.; Dopazo, H.; Gianotti, T.F.; Martino, J.S.; Castaño, G.O.; Pirola, C.J. Mitochondrial Genome Architecture in Non-Alcoholic Fatty Liver Disease: Mitochondrial Genome Mutations and NAFLD. J. Pathol. 2016, 240, 437–449. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The role of autophagy in the heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J. The mouse thermoregulatory system: Its impact on translating biomedical data to humans. Physiol. Behav. 2017, 179, 55–66. [Google Scholar] [CrossRef]
- Meyer, C.W.; Willershäuser, M.; Jastroch, M.; Rourke, B.C.; Fromme, T.; Oelkrug, R.; Heldmaier, G.; Klingenspor, M. Adaptive thermogenesis and thermal conductance in wild-type and UCP1-KO mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1396–R1406. [Google Scholar] [CrossRef] [Green Version]
- Giles, D.A.; Moreno-Fernandez, M.E.; Stankiewicz, T.E.; Graspeuntner, S.; Cappelletti, M.; Wu, D.; Mukherjee, M.; Chan, C.C.; Lawson, M.J.; Klarquist, J.; et al. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex-independent disease modeling. S. Nat. Med. 2017, 23, 829–838. [Google Scholar] [CrossRef]
- Petito, G.; Cioffi, F.; Silvestri, E.; De Matteis, R.; Lattanzi, D.; de Lange, P.; Lombardi, A.; Moreno, M.; Goglia, F.; Lanni, A.; et al. 3,5-Diiodo-L-Thyronine (T2) Administration Affects Visceral Adipose Tissue Inflammatory State in Rats Receiving Long-Lasting High-Fat Diet. Front. Endocrinol. 2021, 12, 703170. [Google Scholar] [CrossRef]
- Nadal-Casellas, A.; Proenza, A.M.; Lladó, I.; Gianotti, M. Sex-dependent differences in rat hepatic lipid accumulation and insulin sensitivity in response to diet-induced obesity. Biochem. Cell Biol. 2012, 90, 164–172. [Google Scholar] [CrossRef]
- Dong, Q.; Kuefner, M.S.; Deng, X.; Bridges, D.; Park, E.A.; Elam, M.B.; Raghow, R. Sex-specific differences in hepatic steatosis in obese spontaneously hypertensive (SHROB) rats. Biol. Sex Differ. 2018, 9, 40. [Google Scholar] [CrossRef]
- Grasselli, E.; Voci, A.; Canesi, L.; De Matteis, R.; Goglia, F.; Cioffi, F.; Fugassa, E.; Gallo, G.; Vergani, L. Direct effects of iodothyronines on excess fat storage in rat hepatocytes. J. Hepatol. 2011, 54, 1230–1236. [Google Scholar] [CrossRef]
- Hsu, S.M.; Raine, L.; Fanger, H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: A comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem. Cytochem. 1981, 29, 577–580. [Google Scholar] [CrossRef] [Green Version]
- Barja, G. Mitochondrial free radical production and aging in mammals and birds. Ann. N. Y. Acad. Sci. 1998, 854, 224–238. [Google Scholar] [CrossRef]
- Silvestri, E.; Lombardi, A.; Coppola, M.; Gentile, A.; Cioffi, F.; Senese, R.; Goglia, F.; Lanni, A.; Moreno, M.; de Lange, P. Differential Effects of 3,5-Diiodo-L-Thyronine and 3,5,3'-Triiodo-L-Thyronine on Mitochondrial Respiratory Pathways in Liver from Hypothyroid Rats. Cell Physiol. Biochem. 2018, 47, 2471–2483. [Google Scholar] [CrossRef]
- Zhou, M.; Diwu, Z.; Panchuk-Voloshina, N.; Haugland, R.P. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: Applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal. Biochem. 1997, 253, 162–168. [Google Scholar] [CrossRef]
- Santos, J.H.; Meyer, J.N.; Mandavilli, B.S.; Van Houten, B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 2006, 314, 183–199. [Google Scholar] [CrossRef] [Green Version]
- Cioffi, F.; Senese, R.; Petito, G.; Lasala, P.; de Lange, P.; Silvestri, E.; Lombardi, A.; Moreno, M.; Goglia, F.; Lanni, A. Both 3,3',5-triiodothyronine and 3,5-diodo-L-thyronine Are Able to Repair Mitochondrial DNA Damage but by Different Mechanisms. Front. Endocrinol. 2019, 10, 216. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Silvestri, E.; Senese, R.; Cioffi, F.; De Matteis, R.; Lattanzi, D.; Lombardi, A.; Giacco, A.; Salzano, A.M.; Scaloni, A.; Ceccarelli, M.; et al. 3,5-Diiodo-L-Thyronine Exerts Metabolically Favorable Effects on Visceral Adipose Tissue of Rats Receiving a High-Fat Diet. Nutrients 2019, 11, 278. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Snel, M.; Jonker, J.T.; Schoones, J.; Lamb, H.; de Roos, A.; Pijl, H.; Smit, J.W.A.; Meinders, A.E.; Jazet, I.M. Ectopic fat and insulin resistance: Pathophysiology and effect of diet and lifestyle interventions. Int. J. Endocrinol. 2012, 2012, 983814. [Google Scholar] [CrossRef]
- Mantena, S.K.; Vaughn, D.P.; Andringa, K.; Eccleston, H.B.; King, A.L.; Abrams, G.A.; Doeller, J.E.; Kraus, D.W.; Darley-Usmar, V.M.; Bailey, S.M. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem. J. 2008, 417, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Ballestri, S.; Zona, S.; Targher, G.; Romagnoli, D.; Baldelli, E.; Nascimbeni, F.; Roverato, A.; Guaraldi, G.; Lonardo, A. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2016, 31, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.I.; Fan, J.G.; Shi, J.P.; Wong, V.W.; Eslam, M.; George, J.; Zheng, M.-H. From NAFLD to MAFLD: A “redefining” moment for fatty liver disease. Chin. Med. J. 2020, 133, 2271–2273. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; George, J. Reply to: Correspondence regarding “A new definition for metabolic dysfunction-associated fatty liver disease: An inter- national expert consensus statement”: Bringing evidence to the NAFLD- MAFLD debate. J. Hepatol. 2020, 73, 1575. [Google Scholar] [CrossRef]
- Sangwung, P.; Petersen, K.F.; Shulman, G.I.; Knowles, J.W. Mitochondrial dysfunction, insulin resistance, and potential genetic implications. Endocrinology 2020, 161, bqaa017. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Shulman, G.I. Therapeutic potential of mitochondrial uncouplers for the treatment of metabolic associated fatty liver disease and NASH. Mol. Metab. 2021, 46, 101178. [Google Scholar] [CrossRef] [PubMed]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef]
- Song, M.; Chen, Y.; Gong, G.; Murphy, E.; Rabinovitch, P.S.; Dorn, G.W. II Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ. Res. 2014, 115, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Mollica, M.P.; Lionetti, L.; Moreno, M.; Lombardi, A.; de Lange, P.; Antonelli, A.; Lanni, A.; Cavaliere, G.; Barletta, A.; Goglia, F. 3,5-diiodo-l-thyronine, by modulating mitochondrial functions, reverses hepatic fat accumulation in rats fed a high-fat diet. J. Hepatol. 2009, 51, 363–370. [Google Scholar] [CrossRef]
- Vial, G.; Dubouchaud, H.; Couturier, K.; Rousselle Cottet, C.; Taleux, N.; Athias, A.; Galinier, A.; Casteilla, L.; Leverve, X.M. Effects of a high-fat diet on energy metabolism and ROS production in rat liver. J. Hepatol. 2011, 54, 348–356. [Google Scholar] [CrossRef]
- Grollman, A.P.; Moriya, M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993, 9, 246–249. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Herbert, K.E.; Lunec, J. Urinary 8-oxo-2'- deoxyguanosine–source, significance and supplements. Free Radic. Res. 2000, 32, 381–397. [Google Scholar] [CrossRef]
- Jones, D.P.; Eklöw, L.; Thor, H.; Orrenius, S. Metabolism of hydrogen peroxide in isolated hepatocytes: Relative contributions of catalase and glutathione peroxidase in decomposition of endogenously generated H2O2. Arch. Biochem. Biophys. 1981, 210, 505–516. [Google Scholar] [CrossRef]
- Verkerk, A.; Jongkind, J.F. Vascular cells under peroxide induced oxidative stress: A balance study on in vitro peroxide handling by vascular endothelial and smooth muscle cells. Free Radic. Res. Commun. 1992, 17, 121–132. [Google Scholar] [CrossRef]
- Hurt, E.M.; Thomas, S.B.; Peng, B.; Farrar, W.L. Molecular consequences of SOD2 expression in epigenetically silenced pancreatic carcinoma cell lines. J. Cancer 2007, 97, 1116–1123. [Google Scholar] [CrossRef]
- Miettinen, M.; Rikala, M.S.; Rys, J.; Lasota, J.; Wang, Z.F. Vascular endothelial growth factor receptor 2 as a marker for malignant vascular tumors and mesothelioma: An immunohistochemical study of 262 vascular endothelial and 1640 nonvascular tumors. Am. J. Surg. Pathol. 2012, 36, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Farge, G.; Falkenberg, M. Organization of DNA in Mammalian Mitochondria. Int. J. Mol. Sci. 2019, 20, 2770. [Google Scholar] [CrossRef] [Green Version]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.G. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef]
- Devasani, K.; Kaul, R.; Majumdar, A. Supplementation of pyrroloquinoline quinone with atorvastatin augments mitochondrial biogenesis and attenuates low grade inflammation in obese rats. Eur. J. Pharmacol. 2020, 15, 173273. [Google Scholar] [CrossRef]
- Chimienti, G.; Orlando, A.; Russo, F.; D’Attoma, B.; Aragno, M.; Aimaretti, E.; Lezza, A.M.S.; Pesce, V. The Mitochondrial Trigger in an Animal Model of Nonalcoholic Fatty Liver Disease. Genes 2021, 12, 1439. [Google Scholar] [CrossRef] [PubMed]
- García-Ruiz, I.; Solís-Muñoz, P.; Fernández-Moreira, D.; Grau, M.; Colina, F.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. High-fat diet decreases activity of the oxidative phosphorylation complexes and causes nonalcoholic steatohepatitis in mice. Dis. Model. Mech. 2014, 7, 1287–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adcock, I.M.; Cosio, B.; Tsaprouni, L.; Barnes, P.J.; Ito, K. Redox regulation of histone deacetylases and glucocorticoid-mediated inhibition of the inflammatory response. Antioxid. Redox Signal. 2005, 7, 144–152. [Google Scholar] [CrossRef]
- Kidane, D.; Chae, W.J.; Czochor, J.; Eckert, K.A.; Glazer, P.M.; Bothwell, A.L.; Sweasy, J.B. Interplay between DNA repair and inflammation, and the link to cancer. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 116–139. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cao, K.; Li, Y.; Zou, X.; Chen, C.; Szeto, I.M.; Dong, Z.; Zhao, Y.; Shi, Y.; Wang, J.; et al. Bitter gourd inhibits the development of obesity-associated fatty liver in C57BL/6 mice fed a high-fat diet. J. Nutr. 2014, 144, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.; Caple, F.; Spiers, A.; Burtle, B.; Daly, A.K.; Williams, E.A.; Hesketh, J.E.; Mathers, J.C. Inter-individual variation in nucleotide excision repair in young adults: Effects of age, adiposity, micronutrient supplementation and genotype. Br. J. Nutr. 2009, 101, 1316–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Roy, M.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division and fusion in metabolism. Curr. Opin. Cell Biol. 2015, 33, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Jiang, L. Dysregulated Mitochondrial Dynamics and Metabolism in Obesity, Diabetes, and Cancer. Front. Endocrinol. 2019, 10, 570. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhang, X.; Han, J.; Man, K.; Zhang, Y.; Chu, E.S.; Nan, Y.; Yu, J. Pro-Inflammatory CXCR3 Impairs Mitochondrial Function in Experimental Non-Alcoholic Steatohepatitis. Theranostics 2017, 7, 4192–4203. [Google Scholar] [CrossRef]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Lin, H.; Xu, Y.; Zhou, F.; Wang, J.; Liu, J.; Zhu, X.; Guo, X.; Tang, Y.; Yao, P. Frataxin-Mediated PINK1-Parkin-Dependent Mitophagy in Hepatic Steatosis: The Protective Effects of Quercetin. Mol. Nutr. Food Res. 2018, 62, e1800164. [Google Scholar] [CrossRef]
- Zhou, T.; Chang, L.; Luo, Y.; Zhou, Y.; Zhang, J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Boil. 2019, 21, 101120. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 2015, 61, 486–496. [Google Scholar] [CrossRef] [Green Version]
- Edmunds, L.R.; Xie, B.; Mills, A.M.; Huckestein, B.R.; Undamatla, R.; Murali, A.; Pangburn, M.M.; Martin, J.; Sipula, I.; Kaufman, B.A.; et al. Liver-specific Prkn knockout mice are more susceptible to diet-induced hepatic steatosis and insulin resistance. Mol. Metab. 2020, 41, 101051. [Google Scholar] [CrossRef]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring in vivo mitophagy. Mol. Cell 2015, 60, e685–e696. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.W.; Cannon, B.; Nedergaard, J. Optimal housing temperatures for mice to mimic the thermal environment of humans: An experimental study. Mol. Metab. 2018, 7, 161–170. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | N | HFD |
|---|---|---|
| Bodyweight gain (g) | 115 ± 12.65 | 192.30 ± 8.18 * |
| Liver weight (g) | 7.48 ± 0.23 | 9.59 ± 0.36 * |
| Liver weight (g)/ Bodyweight (g) | 0.021 ± 0.00029 | 0.022 ± 0.00032 |
| WAT weight (g) | 11.20 ± 0.98 | 31.70 ± 1.80 * |
| WAT weight (g)/ Bodyweight | 0.030 ± 0.0015 | 0.074 ± 0.0023 * |
| Energy intake (KJ) | 5098 ± 408 | 6798 ± 498 * |
| Respiratory quotient | 0.99 ± 0.058 | 0.72 ± 0.013 * |
| Triglycerides (mg/dL) | 115 ± 5.45 | 260.50 ± 17.33 * |
| Cholesterol (mg/dL) | 39 ± 4.39 | 74.70 ± 3.11 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cioffi, F.; Giacco, A.; Petito, G.; de Matteis, R.; Senese, R.; Lombardi, A.; de Lange, P.; Moreno, M.; Goglia, F.; Lanni, A.; et al. Altered Mitochondrial Quality Control in Rats with Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD) Induced by High-Fat Feeding. Genes 2022, 13, 315. https://doi.org/10.3390/genes13020315
Cioffi F, Giacco A, Petito G, de Matteis R, Senese R, Lombardi A, de Lange P, Moreno M, Goglia F, Lanni A, et al. Altered Mitochondrial Quality Control in Rats with Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD) Induced by High-Fat Feeding. Genes. 2022; 13(2):315. https://doi.org/10.3390/genes13020315
Chicago/Turabian StyleCioffi, Federica, Antonia Giacco, Giuseppe Petito, Rita de Matteis, Rosalba Senese, Assunta Lombardi, Pieter de Lange, Maria Moreno, Fernando Goglia, Antonia Lanni, and et al. 2022. "Altered Mitochondrial Quality Control in Rats with Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD) Induced by High-Fat Feeding" Genes 13, no. 2: 315. https://doi.org/10.3390/genes13020315