
GUCY2D-Related Retinal Dystrophy with Autosomal Dominant Inheritance—A Multicenter Case Series and Review of Reported Data

 and
and 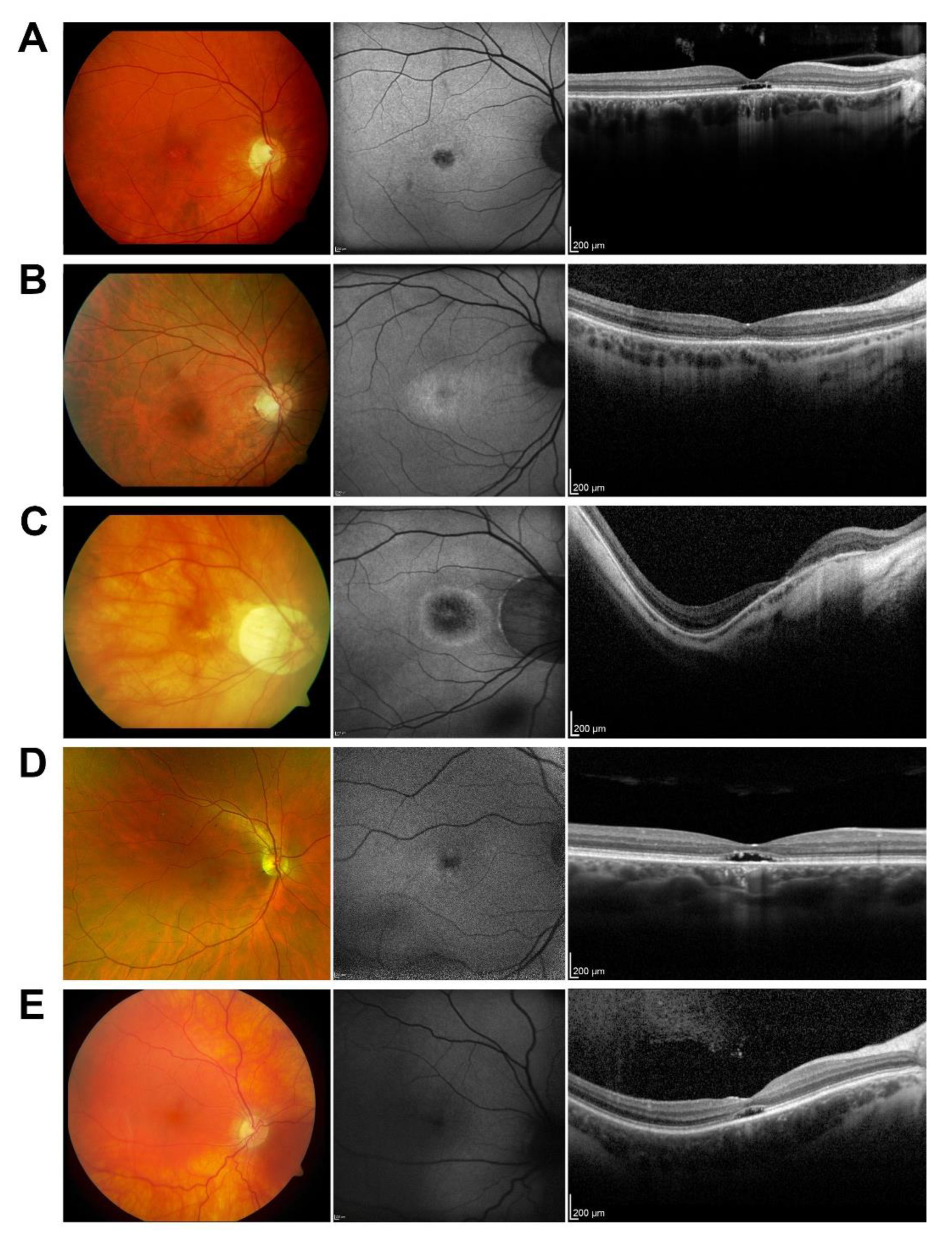{kind=link}
{kind=link}
{kind=link}
{kind=link}
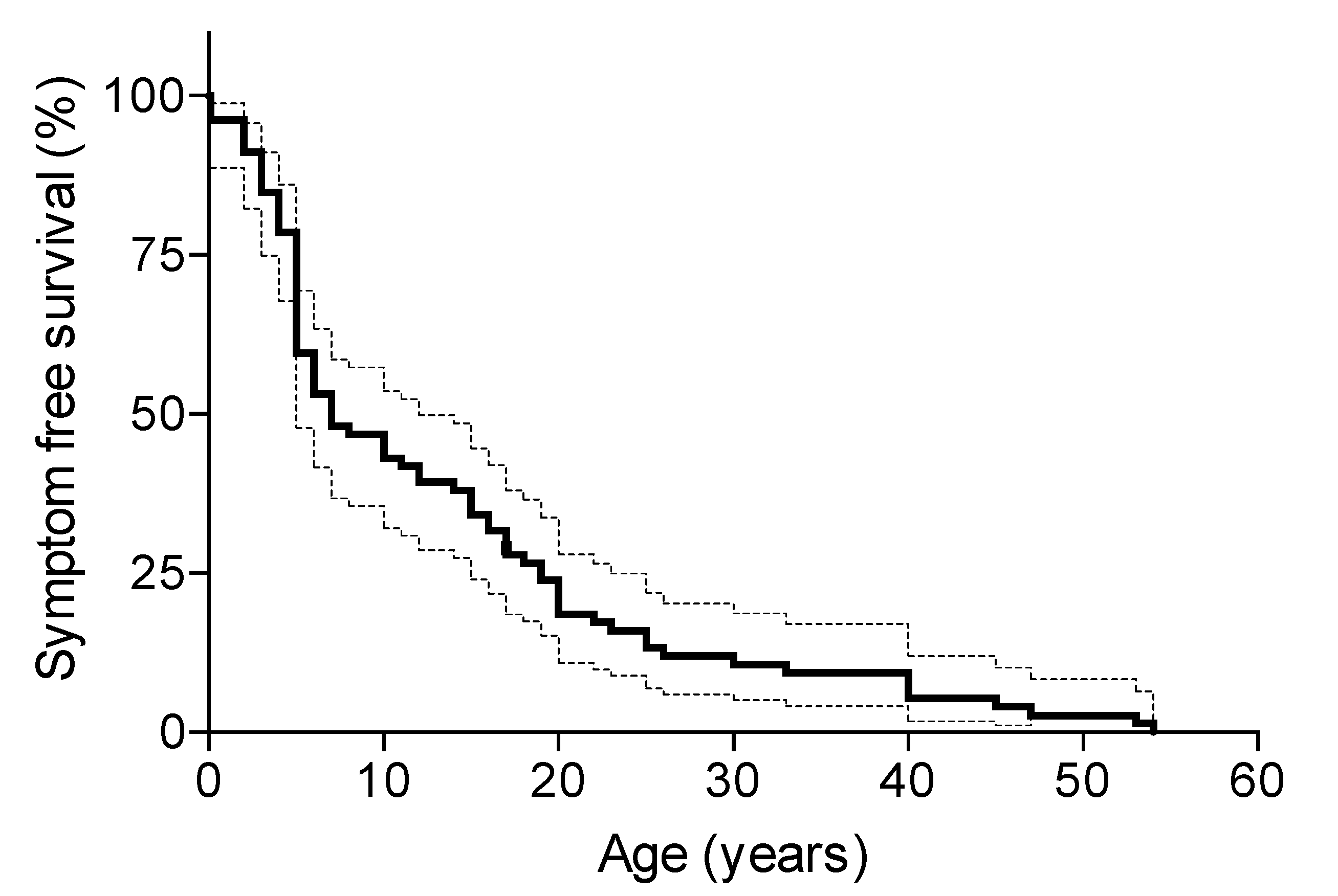{kind=link}
Abstract
:1. Introduction
2. Methods
3. Results
3.1. Patient Cohort Characteristics
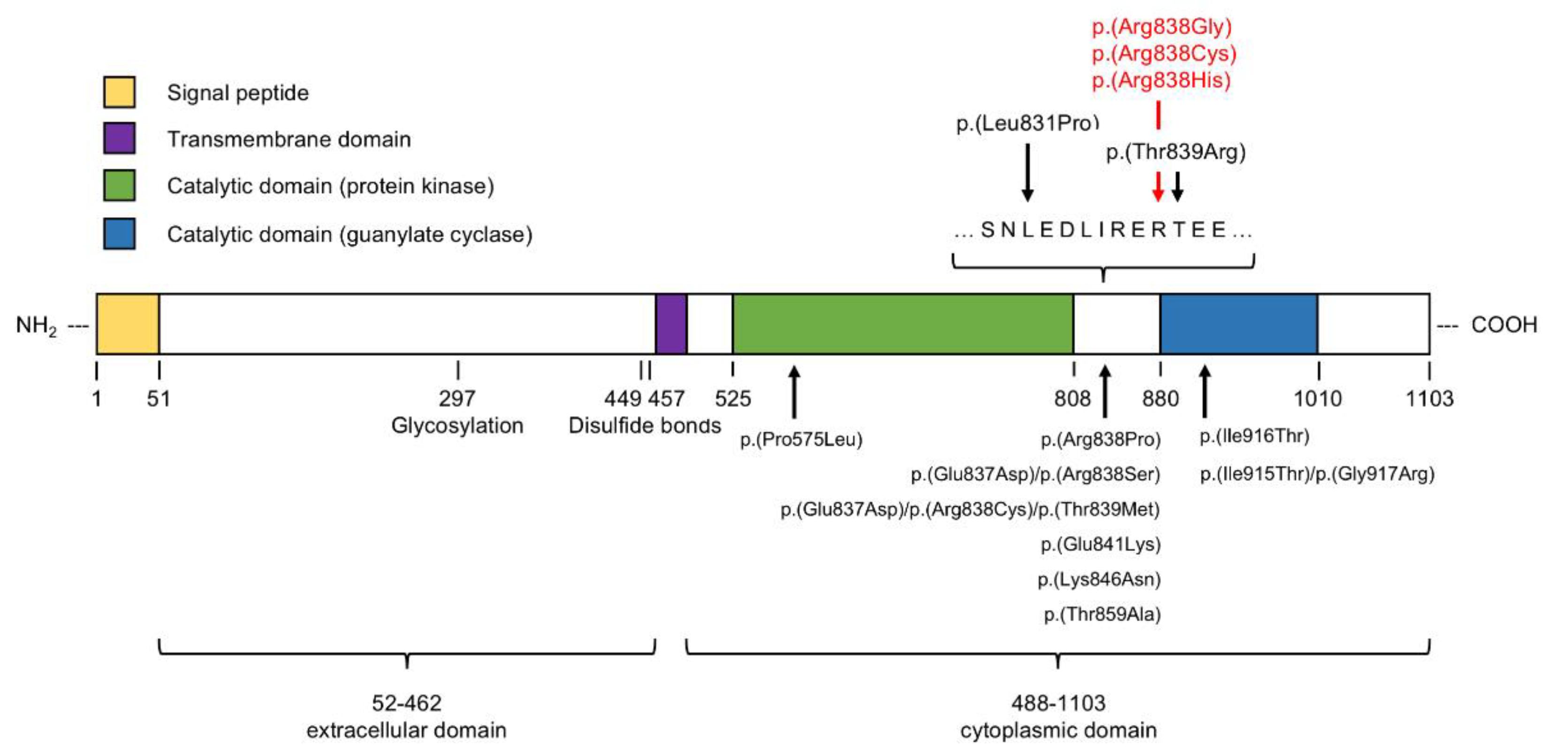
3.2. Molecular Assessment
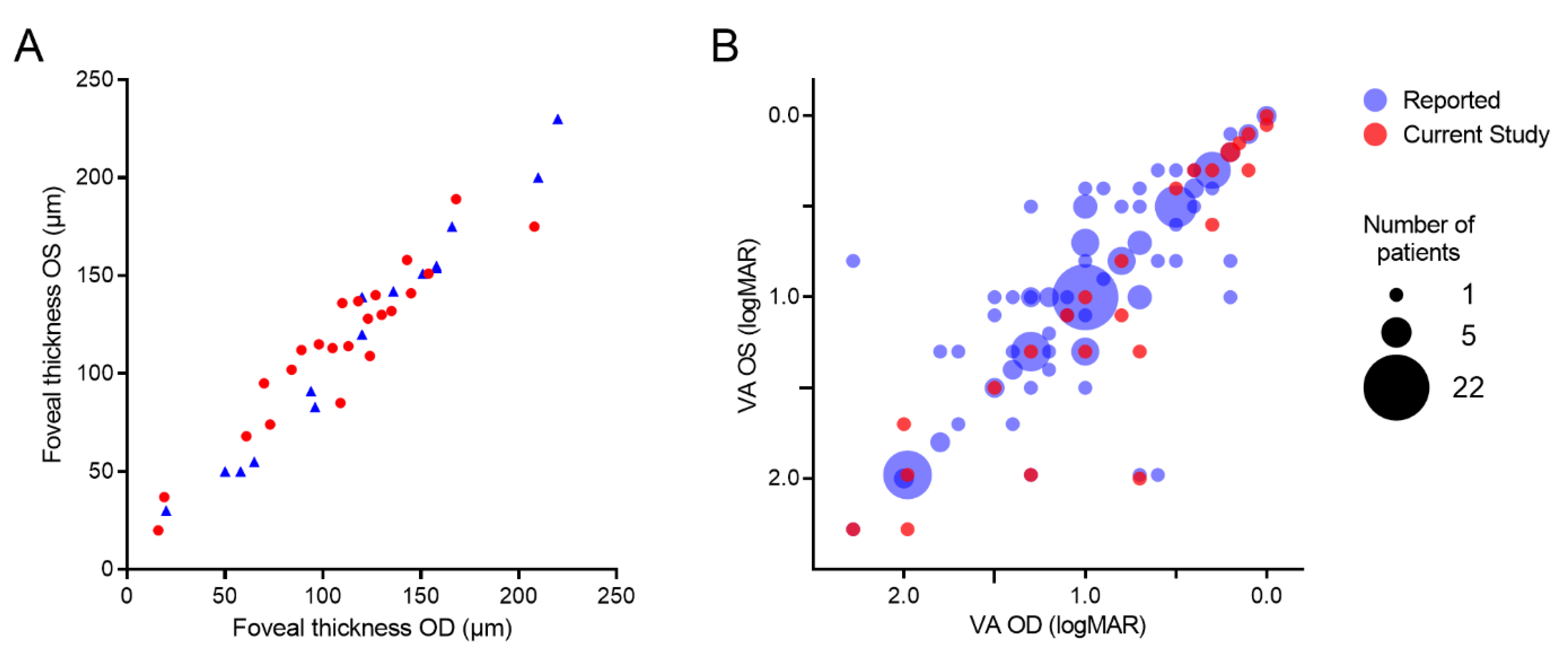
3.3. Disease Symmetry between Eyes
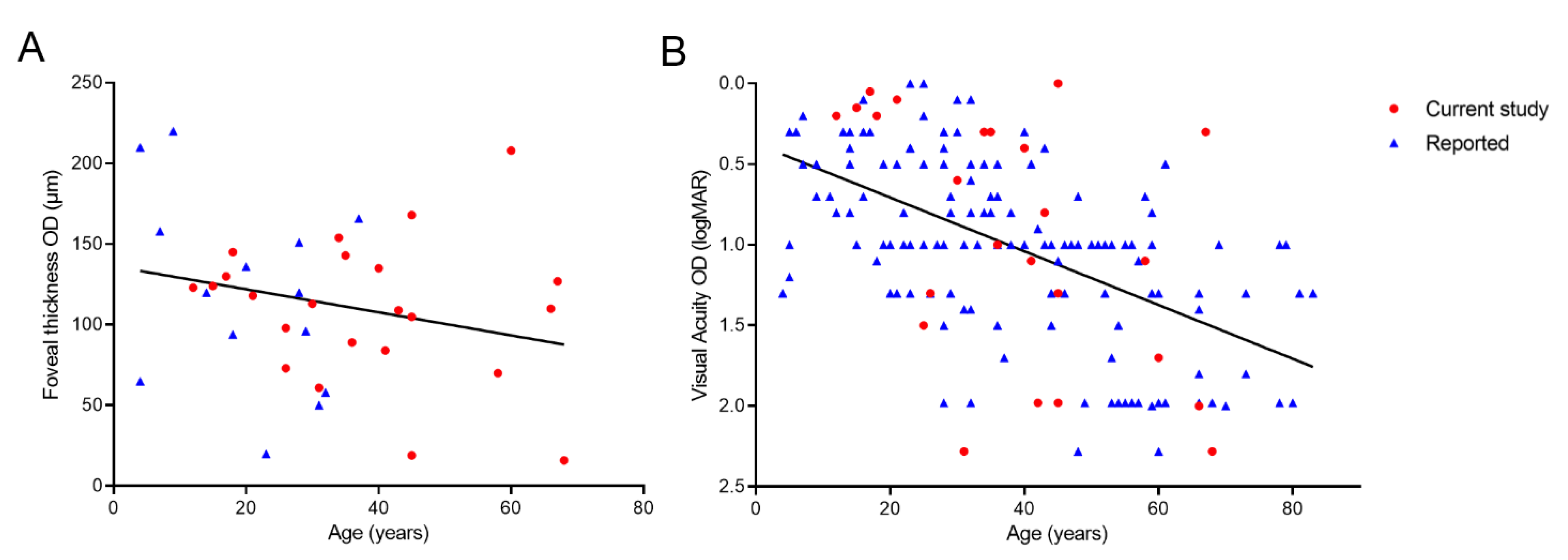
3.4. Disease Progression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- RetNet. Summaries of Genes and Loci Causing Retinal Disease. Available online: https://sph.uth.edu/retnet/sum-dis.htm (accessed on 16 February 2020).
- Hamel, C.P. Cone Rod Dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef] [Green Version]
- Birtel, J.; Yusuf, I.H.; Priglinger, C.; Rudolph, G.; Charbel Issa, P. Diagnosis of Inherited Retinal Diseases. Klin. Monbl. Augenheilkd 2021, 238, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Kurz-Levin, M.M.; Halfyard, A.S.; Bunce, C.; Bird, A.C.; Holder, G.E. Clinical Variations in Assessment of Bull’s-Eye Maculopathy. Arch Ophthalmol. 2002, 120, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasser, F.; Kurtenbach, A.; Kohl, S.; Obermaier, C.; Stingl, K.; Zrenner, E. Retinal dystrophies with bull’s-eye maculopathy along with negative ERGs. Doc. Ophthalmol. 2019, 139, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive Cone and Cone-Rod Dystrophies: Clinical Features, Molecular Genetics and Prospects for Therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.; Scheetz, T.; Sheffield, V.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Müller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef]
- Hanein, S.; Perrault, I.; Gerber, S.; Tanguy, G.; Barbet, F.; Ducroq, D.; Calvas, P.; Dollfus, H.; Hamel, C.; Lopponen, T.; et al. Leber Congenital Amaurosis: Comprehensive Survey of the Genetic Heterogeneity, Refinement of the Clinical Definition, and Genotype-Phenotype Correlations as a Strategy for Molecular Diagnosis. Hum. Mutat. 2004, 23, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Sharon, D.; Wimberg, H.; Kinarty, Y.; Koch, K.-W. Genotype-functional-phenotype correlations in photoreceptor guanylate cyclase (GC-E) encoded by GUCY2D. Prog. Retin. Eye Res. 2018, 63, 69–91. [Google Scholar] [CrossRef]
- Zobor, D.; Zrenner, E.; Wissinger, B.; Kohl, S.; Jagle, H. Gucy2d- or Guca1a-Related Autosomal Dominant Cone-Rod Dystrophy: Is There a Phenotypic Difference? Retina 2014, 34, 1576–1587. [Google Scholar] [CrossRef]
- Went, L.N.; van Schooneveld, M.J.; Oosterhuis, J.A. Late onset dominant cone dystrophy with early blue cone involvement. J. Med Genet. 1992, 29, 295–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsokolas, G.; Almuhtaseb, H.; Griffiths, H.; Shawkat, F.; Pengelly, R.; Ennis, S.; Lotery, A. Long term follow-up of a family with GUCY2D dominant cone dystrophy. Int. J. Ophthalmol. 2018, 11, 1945–1950. [Google Scholar] [CrossRef] [PubMed]
- Oishi, M.; Oishi, A.; Gotoh, N.; Ogino, K.; Higasa, K.; Iida, K.; Makiyama, Y.; Morooka, S.; Matsuda, F.; Yoshimura, N. Next-generation sequencing-based comprehensive molecular analysis of 43 Japanese patients with cone and cone-rod dystrophies. Mol. Vis. 2016, 22, 150–160. [Google Scholar] [PubMed]
- Jiang, F.; Xu, K.; Zhang, X.; Xie, Y.; Bai, F.; Li, Y. GUCY2D mutations in a Chinese cohort with autosomal dominant cone or cone–rod dystrophies. Doc. Ophthalmol. 2015, 131, 105–114. [Google Scholar] [CrossRef]
- Lazar, C.H.; Mutsuddi, M.; Kimchi, A.; Zelinger, L.; Mizrahi-Meissonnier, L.; Marks-Ohana, D.; Boleda, A.; Ratnapriya, R.; Sharon, D.; Swaroop, A.; et al. Whole Exome Sequencing Reveals GUCY2D as a Major Gene Associated With Cone and Cone-Rod Dystrophy in Israel. Investig. Opthalmol. Vis. Sci. 2014, 56, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, R.; Robson, A.; Holder, G.E.; Stockman, A.; Egan, C.; Moore, A.T.; Webster, A.R. A detailed phenotypic description of autosomal dominant cone dystrophy due to a de novo mutation in the GUCY2D gene. Eye 2014, 28, 481–487. [Google Scholar] [CrossRef]
- Zhao, X.; Ren, Y.; Zhang, X.; Chen, C.; Dong, B.; Li, Y. A novel GUCY2D mutation in a Chinese family with dominant cone dystrophy. Mol. Vis. 2013, 19, 1039–1046. [Google Scholar]
- Garcia-Hoyos, M.; Auz-Alexandre, C.L.; Almoguera, B.; Cantalapiedra, D.; Riveiro-Alvarez, R.; Lopez-Martinez, M.A.; Gimenez, A.; Blanco-Kelly, F.; Avila-Fernandez, A.; Trujillo-Tiebas, M.J.; et al. Mutation Analysis at Codon 838 of the Guanylate Cyclase 2d Gene in Spanish Families with Autosomal Dominant Cone, Cone-Rod, and Macular Dystrophies. Mol. Vis. 2011, 17, 1103–1109. [Google Scholar]
- Kim, B.J.; Ibrahim, M.A.; Goldberg, M.F. Use of Spectral Domain-Optical Coherence Tomography to Visualize Photoreceptor Abnormalities in Cone-Rod Dystrophy 6. Retin Cases Brief Rep. Retin. Cases Brief Rep. 2011, 5, 56–61. [Google Scholar] [CrossRef]
- Kitiratschky, V.B.; Wilke, R.; Renner, A.B.; Kellner, U.; Vadala, M.; Birch, D.G.; Wissinger, B.; Zrenner, E.; Kohl, S. Mutation Analysis Identifies Gucy2d as the Major Gene Responsible for Autosomal Dominant Progressive Cone Degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 5015–5023. [Google Scholar] [CrossRef] [Green Version]
- Small, K.W.; Silva-Garcia, R.; Udar, N.; Nguyen, E.V.; Heckenlively, J.R. New Mutation, P575L, in the GUCY2D Gene in a Family With Autosomal Dominant Progressive Cone Degeneration. Arch. Ophthalmol. 2008, 126, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.; Whittock, N.; Searle, A.; Croft, M.; Brewer, C.; Cole, M. Phenotype of autosomal dominant cone–rod dystrophy due to the R838C mutation of the GUCY2D gene encoding retinal guanylate cyclase-1. Eye 2006, 21, 1220–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Nakamura, M.; Ohnishi, Y.; Miyake, Y. Autosomal Dominant Cone-Rod Dystrophy with R838H and R838C Mutations in the GUCY2D Gene in Japanese Patients. Jpn. J. Ophthalmol. 2004, 48, 228–235. [Google Scholar] [CrossRef]
- Ito, S.; Nakamura, M.; Nuno, Y.; Ohnishi, Y.; Nishida, T.; Miyake, Y. Novel complex GUCY2D mutation in Japanese family with cone-rod dystrophy. Investig. Opthalmol. Vis. Sci. 2004, 45, 1480–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downes, S.M.; Payne, A.; Kelsell, R.E.; Fitzke, F.W.; Holder, G.E.; Hunt, D.; Moore, A.T.; Bird, A.C. Autosomal Dominant Cone-Rod Dystrophy With Mutations in the Guanylate Cyclase 2D Gene Encoding Retinal Guanylate Cyclase-1. Arch. Ophthalmology 2001, 119, 1667–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory-Evans, K.; Kelsell, R.E.; Gregory-Evans, C.Y.; Downes, S.M.; Fitzke, F.W.; Holder, G.E.; Simunovic, M.; Mollon, J.D.; Taylor, R.; Hunt, D.M.; et al. Autosomal Dominant Cone-Rod Retinal Dystrophy (Cord6) from Heterozygous Mutation of Gucy2d, Which Encodes Retinal Guanylate Cyclase. Ophthalmology 2000, 107, 55–61. [Google Scholar] [CrossRef]
- Huang, L.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Sun, W.; Xu, Y.; Xin, W.; Guo, X.; Zhang, Q. Molecular genetics of cone-rod dystrophy in Chinese patients: New data from 61 probands and mutation overview of 163 probands. Exp. Eye Res. 2016, 146, 252–258. [Google Scholar] [CrossRef]
- Huang, L.; Li, S.; Xiao, X.; Jia, X.; Wang, P.; Guo, X.; Zhang, Q. Screening for variants in 20 genes in 130 unrelated patients with cone-rod dystrophy. Mol. Med. Rep. 2013, 7, 1779–1785. [Google Scholar] [CrossRef]
- Xiao, X.; Guo, X.; Jia, X.; Li, S.; Wang, P.; Zhang, Q. A recurrent mutation in GUCY2D associated with autosomal dominant cone dystrophy in a Chinese family. Mol. Vis. 2011, 17, 3271–3278. [Google Scholar]
- Van Ghelue, M.; Eriksen, H.L.; Ponjavic, V.; Fagerheim, T.; Andreasson, S.; Forsman-Semb, K.; Sandgren, O.; Holmgren, G.; Tranebjaerg, L. Autosomal Dominant Cone-Rod Dystrophy Due to a Missense Mutation (R838c) in the Guanylate Cyclase 2d Gene (Gucy2d) with Preserved Rod Function in One Branch of the Family. Ophthalmic Genet. 2000, 21, 197–209. [Google Scholar] [CrossRef]
- Perrault, I.; Rozet, J.-M.; Gerber, S.; Kelsell, R.E.; Souied, E.; Cabot, A.; Hunt, D.M.; Munnich, A.; Kaplan, J. A retGC-1 Mutation in Autosomal Dominant Cone-Rod Dystrophy. Am. J. Hum. Genet. 1998, 63, 651–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Castro-Miro, M.; Pomares, E.; Lorés-Motta, L.; Tonda, R.; Dopazo, J.; Marfany, G.; Gonzàlez-Duarte, R. Combined Genetic and High-Throughput Strategies for Molecular Diagnosis of Inherited Retinal Dystrophies. PLoS ONE 2014, 9, e88410. [Google Scholar] [CrossRef]
- McCullough, K.T.; Boye, S.L.; Fajardo, D.; Calabro, K.; Peterson, J.J.; Strang, C.E.; Chakraborty, D.; Gloskowski, S.; Haskett, S.; Samuelsson, S.; et al. Somatic Gene Editing of GUCY2D by AAV-CRISPR/Cas9 Alters Retinal Structure and Function in Mouse and Macaque. Hum. Gene Ther. 2019, 30, 571–589. [Google Scholar] [CrossRef]
- Cooper, D.N.; Youssoufian, H. The CpG dinucleotide and human genetic disease. Qual. Life Res. 1988, 78, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.M.; Morris, A.G.; Downes, S.M.; Johnson, S.; Bird, A.C.; Moore, A.T.; Bhattacharya, S.S.; Hunt, D.M. Clustering and frequency of mutations in the retinal guanylate cyclase (GUCY2D) gene in patients with dominant cone-rod dystrophies. J. Med. Genet. 2001, 38, 611–614. [Google Scholar] [CrossRef]
- Wilkie, S.E.; Newbold, R.J.; Deery, E.; Walker, C.; Stinton, I.; Ramamurthy, V.; Hurley, J.B.; Bhattacharya, S.S.; Warren, M.J.; Hunt, D.M. Functional characterization of missense mutations at codon 838 in retinal guanylate cyclase correlates with disease severity in patients with autosomal dominant cone-rod dystrophy. Hum. Mol. Genet. 2000, 9, 3065–3073. [Google Scholar] [CrossRef]
- Tucker, C.; Woodcock, S.C.; Kelsell, R.E.; Ramamurthy, V.; Hunt, D.; Hurley, J.B. Biochemical analysis of a dimerization domain mutation in RetGC-1 associated with dominant cone-rod dystrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 9039–9044. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Reed, J.C. Mitochondria and Apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Liu, X.; Seno, K.; Nishizawa, Y.; Hayashi, F.; Yamazaki, A.; Matsumoto, H.; Wakabayashi, T.; Usukura, J. Ultrastructural localization of retinal guanylate cyclase in human and monkey retinas. Exp. Eye Res. 1994, 59, 761–768. [Google Scholar] [CrossRef]
- Jespersgaard, C.; Fang, M.; Bertelsen, M.; Dang, X.; Jensen, H.; Chen, Y.; Bech, N.; Dai, L.; Rosenberg, T.; Zhang, J.; et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and Safety of Voretigene Neparvovec (Aav2-Hrpe65v2) in Patients with Rpe65-Mediated Inherited Retinal Dystrophy: A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neubauer, J.; Hahn, L.; Birtel, J.; Boon, C.J.F.; Charbel Issa, P.; Fischer, M.D. GUCY2D-Related Retinal Dystrophy with Autosomal Dominant Inheritance—A Multicenter Case Series and Review of Reported Data. Genes 2022, 13, 313. https://doi.org/10.3390/genes13020313
Neubauer J, Hahn L, Birtel J, Boon CJF, Charbel Issa P, Fischer MD. GUCY2D-Related Retinal Dystrophy with Autosomal Dominant Inheritance—A Multicenter Case Series and Review of Reported Data. Genes. 2022; 13(2):313. https://doi.org/10.3390/genes13020313
Chicago/Turabian StyleNeubauer, Jonas, Leo Hahn, Johannes Birtel, Camiel J. F. Boon, Peter Charbel Issa, and M. Dominik Fischer. 2022. "GUCY2D-Related Retinal Dystrophy with Autosomal Dominant Inheritance—A Multicenter Case Series and Review of Reported Data" Genes 13, no. 2: 313. https://doi.org/10.3390/genes13020313