
Identification of a Four-Gene Signature Associated with the Prognosis Prediction of Lung Adenocarcinoma Based on Integrated Bioinformatics Analysis

 and
and
Abstract
:1. Introduction
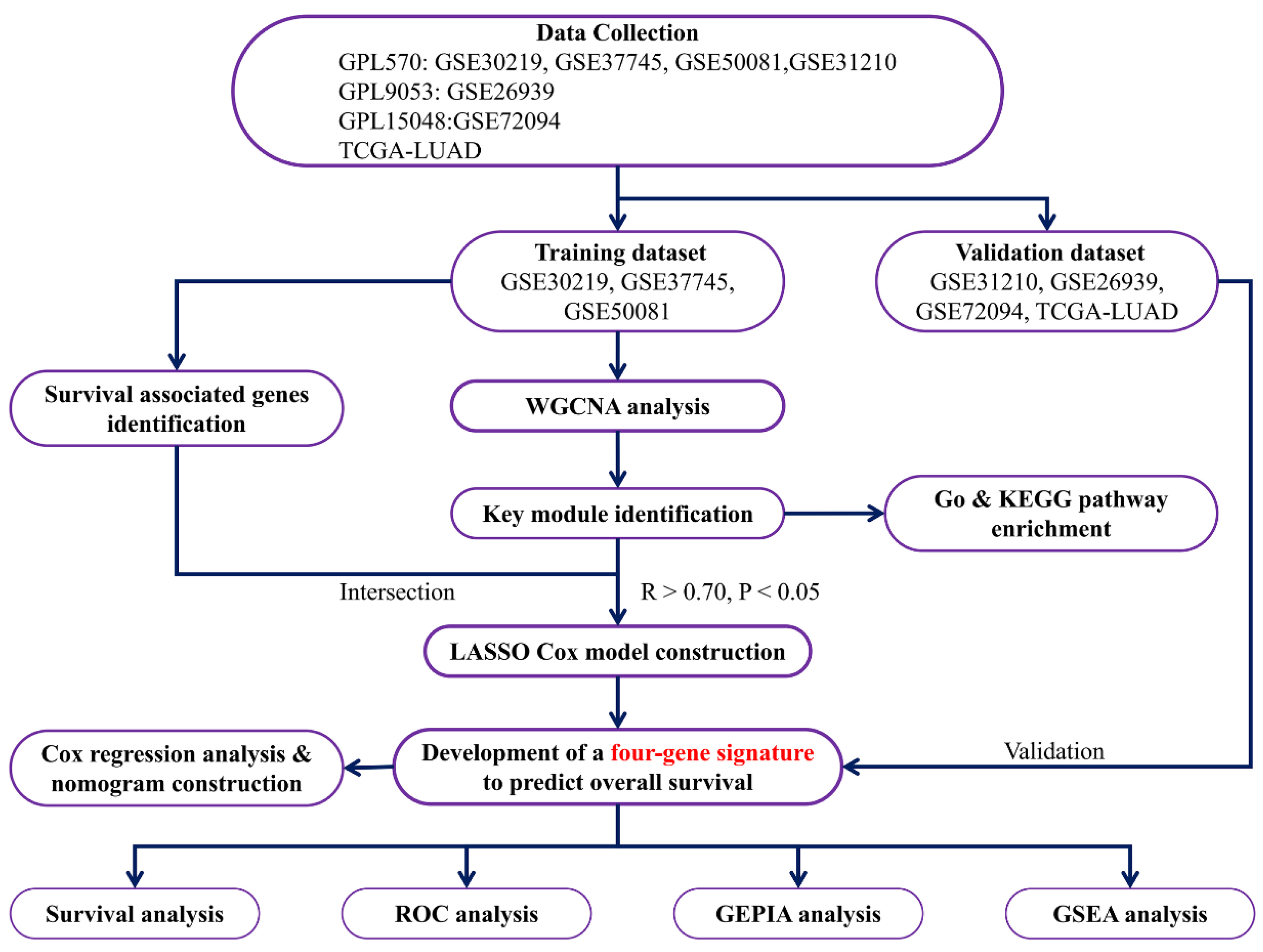
2. Materials and Methods
2.1. Data-Collection and Pre-Processing
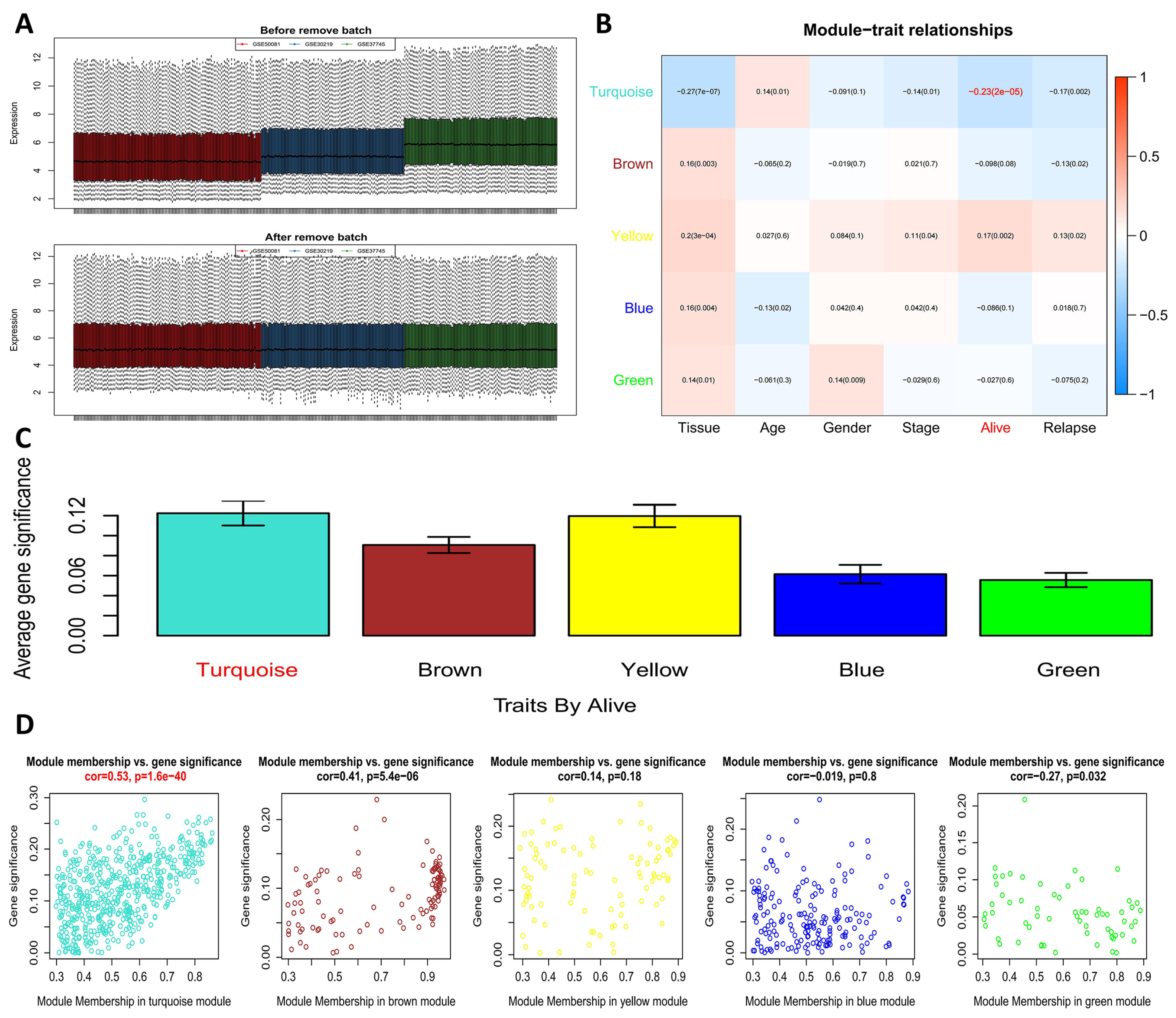
2.2. WGCNA to Screen Out a Key Module and Genes Related to Survival
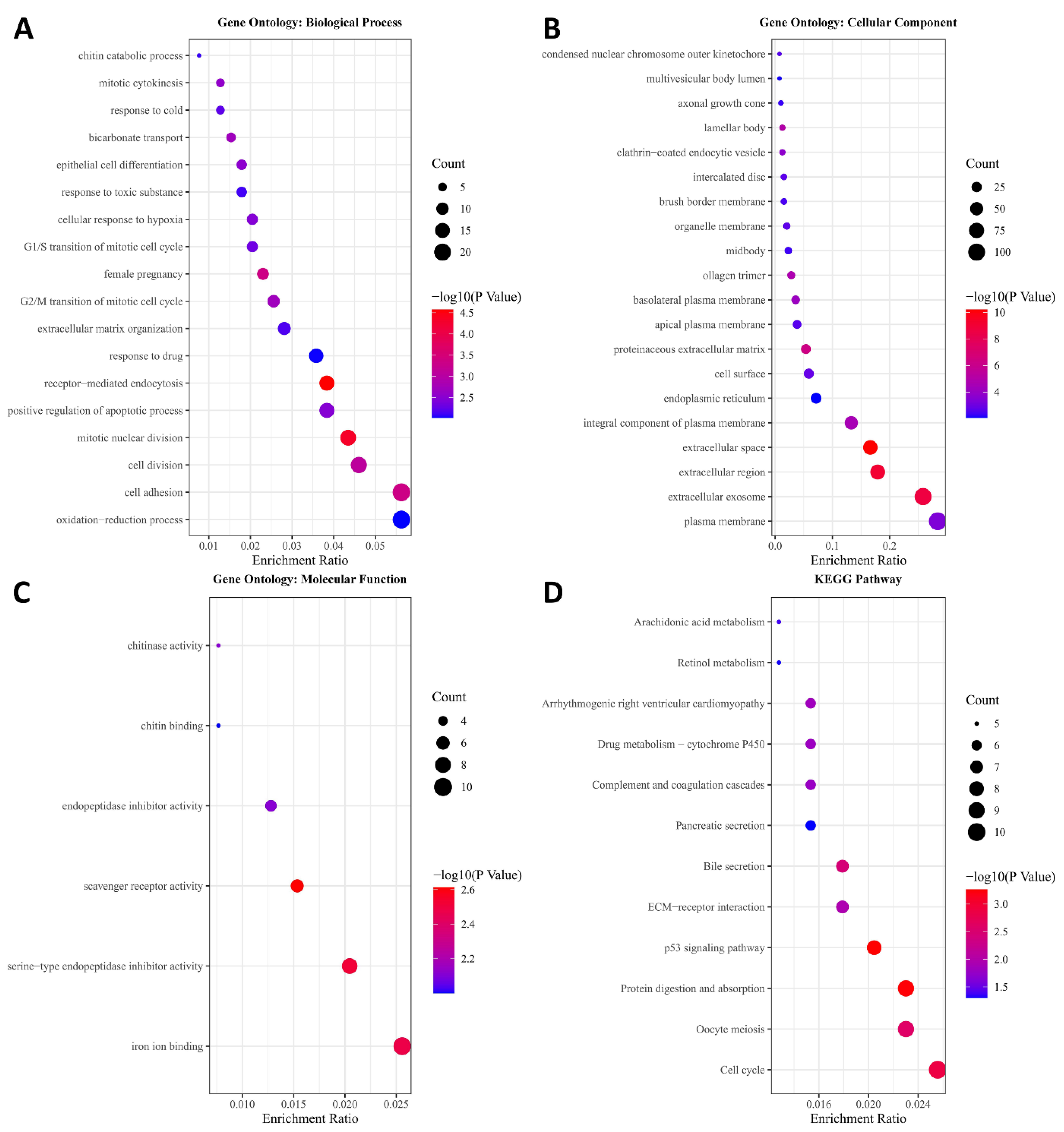
2.3. GO Enrichment and KEGG Pathway Analysis
2.4. Prognostic Genes Screening Associated with OS and DFS
2.5. Prognostic Gene Signature Construction and Validation
2.6. Univariate and Multivariate Cox Regression Analysis
2.7. Gene Set Enrichment Analysis (GSEA)
2.8. Cell Apoptosis Assay
3. Results
3.1. Key Module Identification and Functional Enrichment Analysis
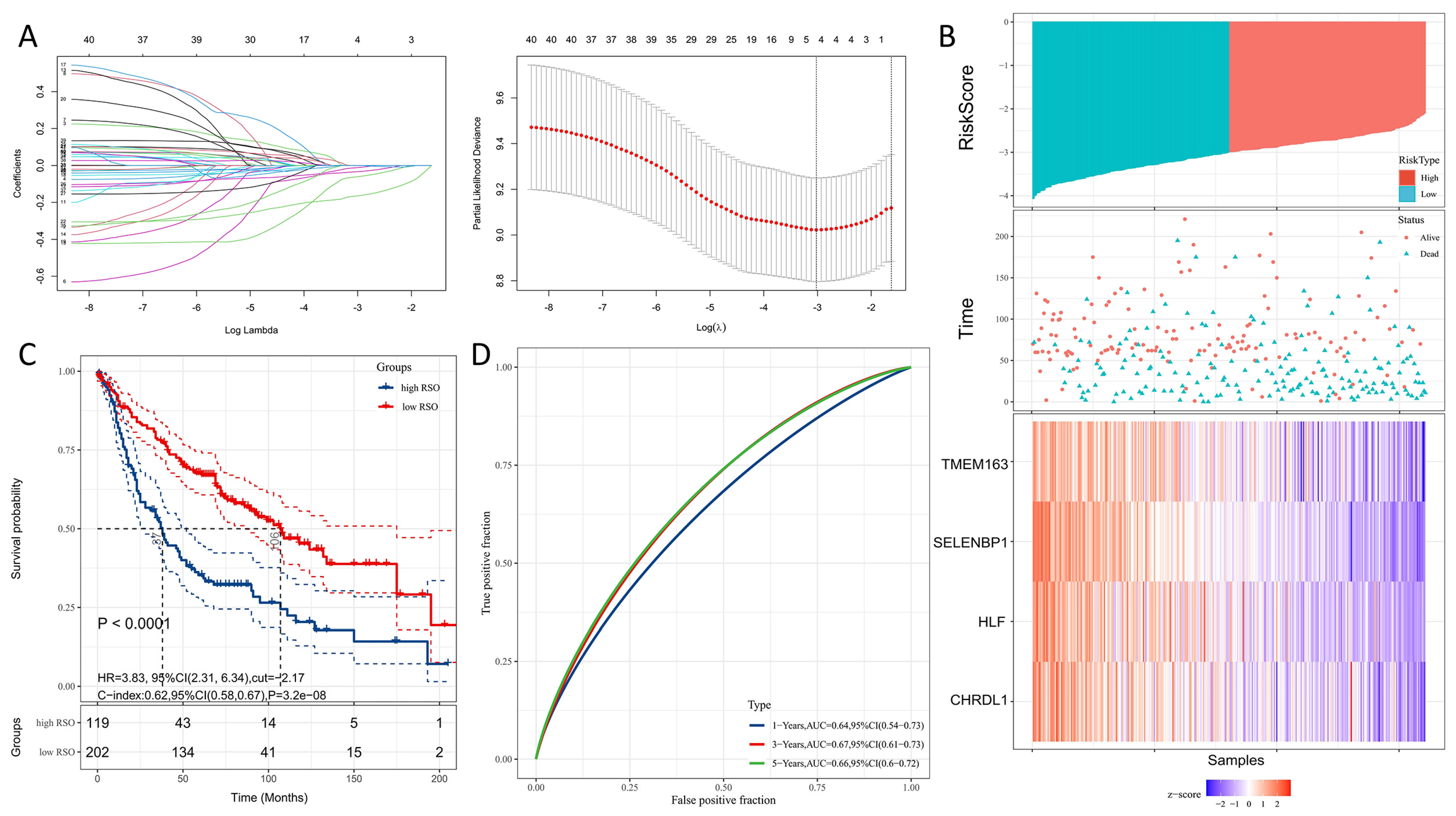
3.2. Modeling Gene Identification and Construction of a Four-Gene Signature for Predicting OS
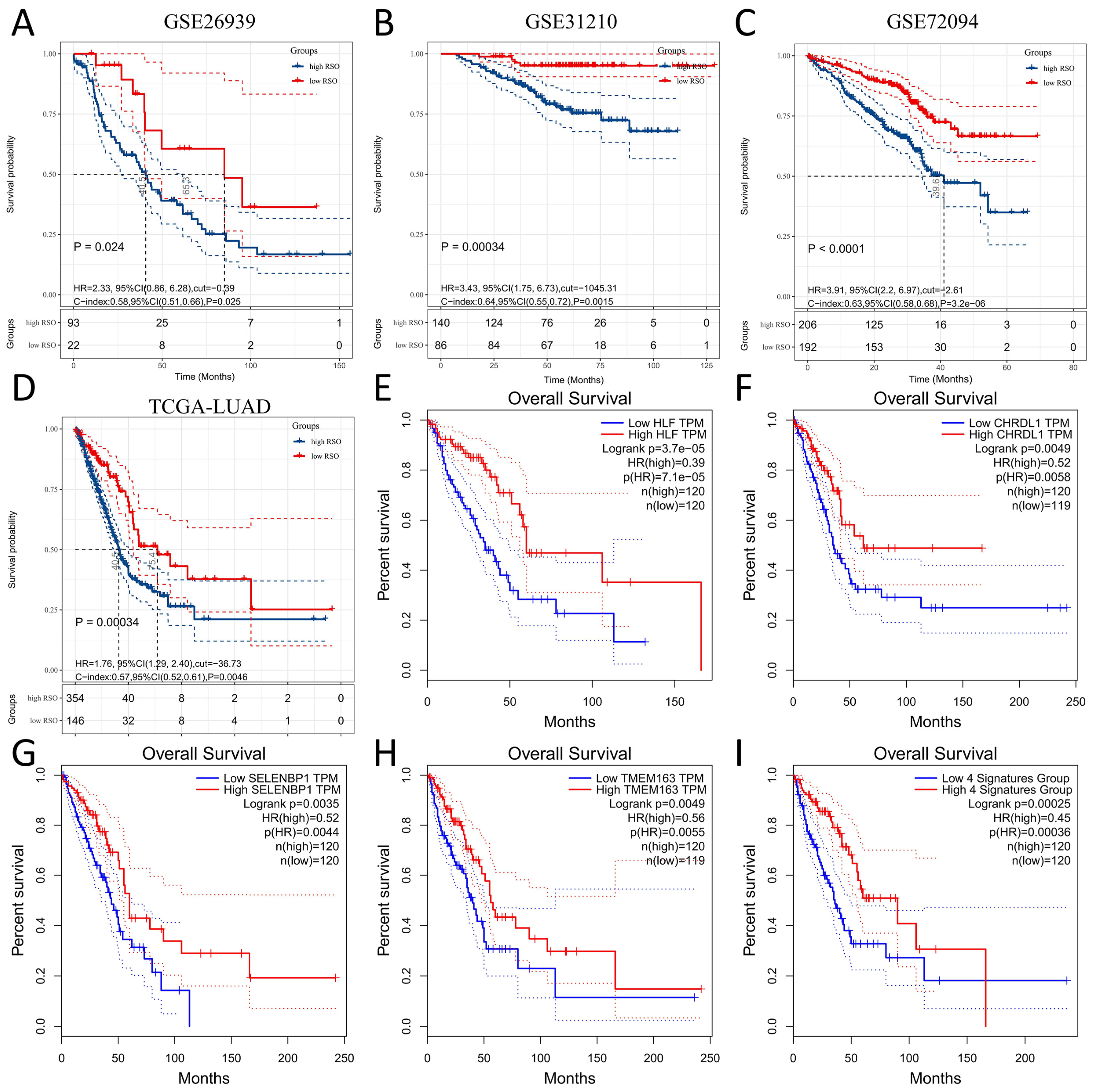
3.3. Prognostic Value of the Four-Gene Signature
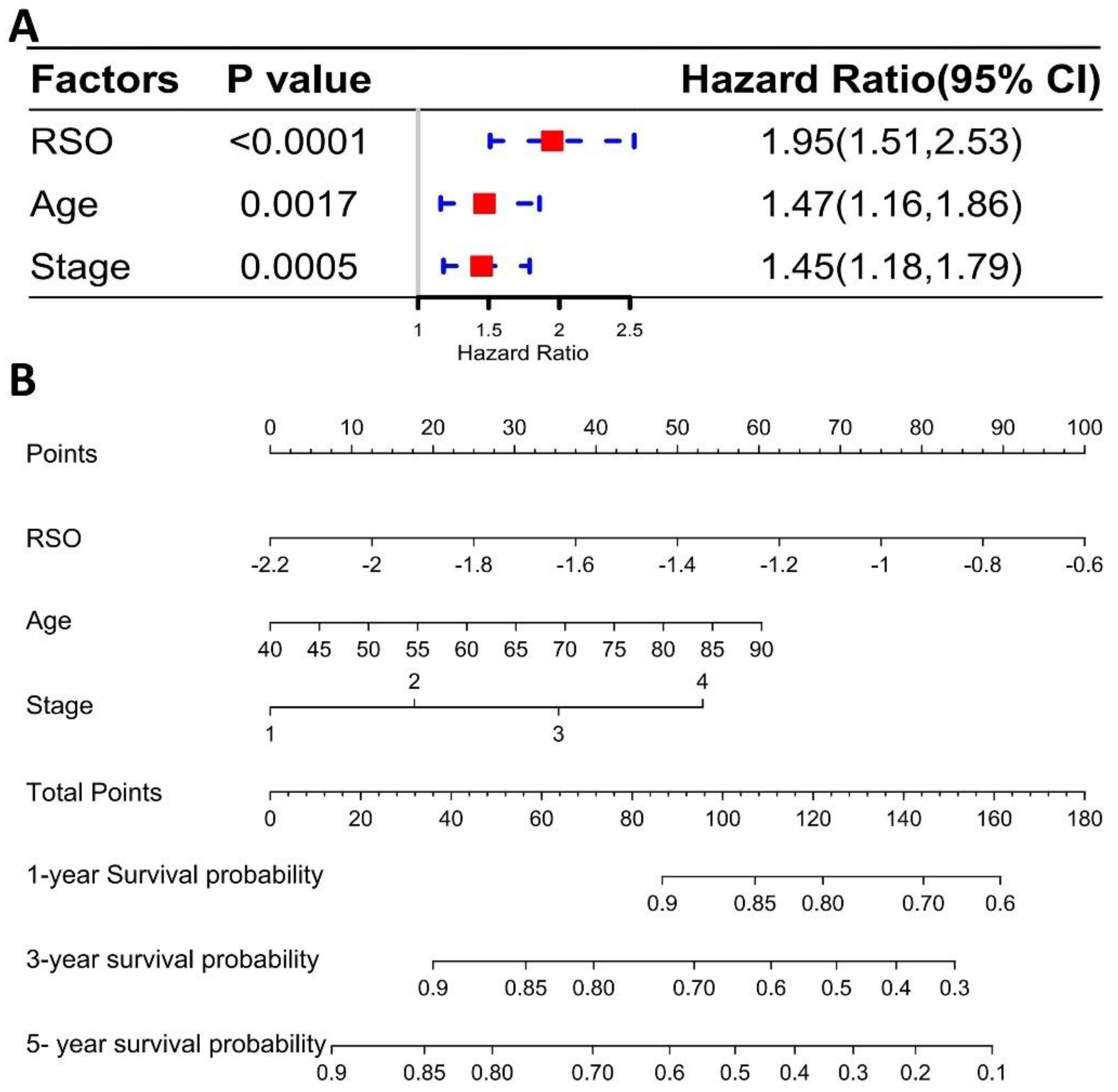
3.4. The Four-Gene Signature Could Be a Better Prognostic Factor Than Clinical Factors in the Training Set
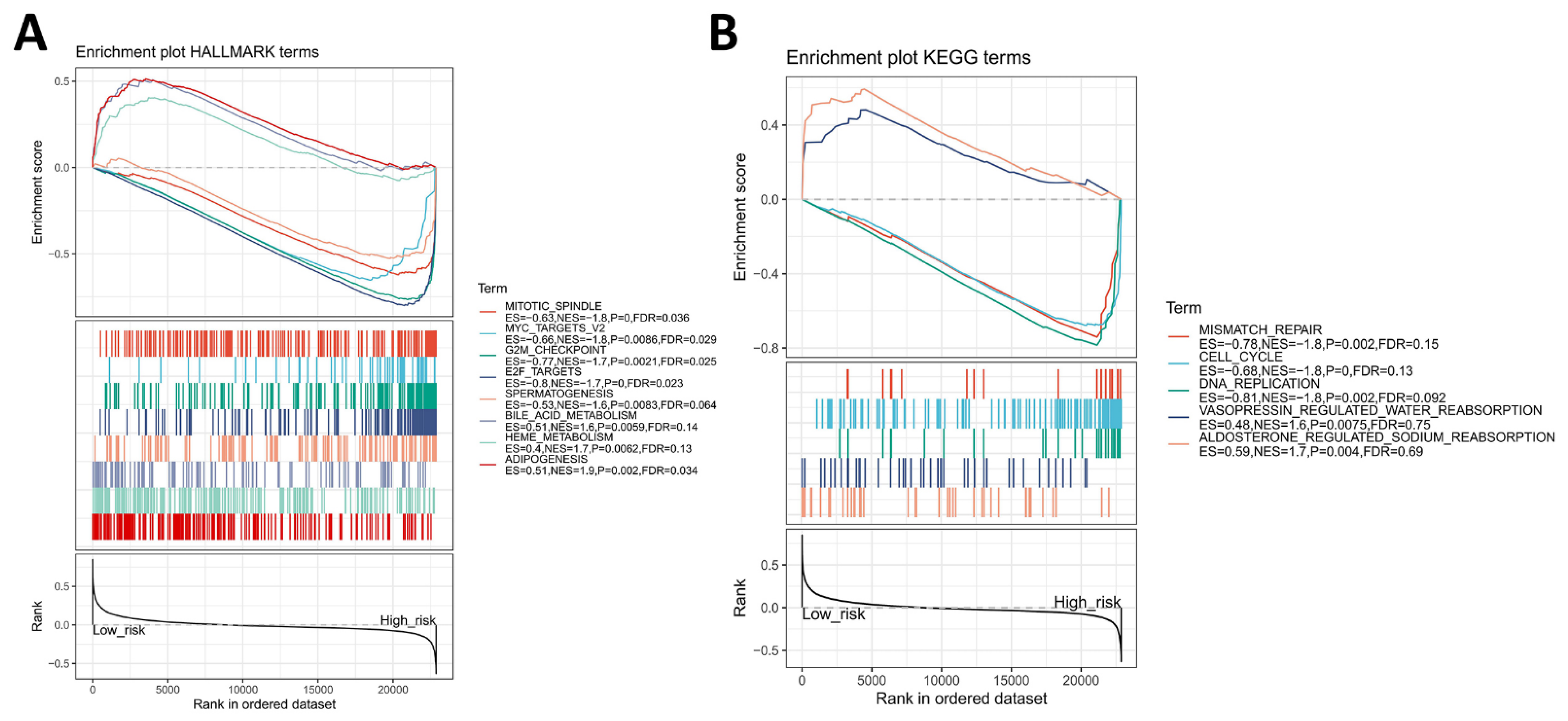
3.5. Identification of Four-Gene Signature Associated Hallmark and KEGG Pathway
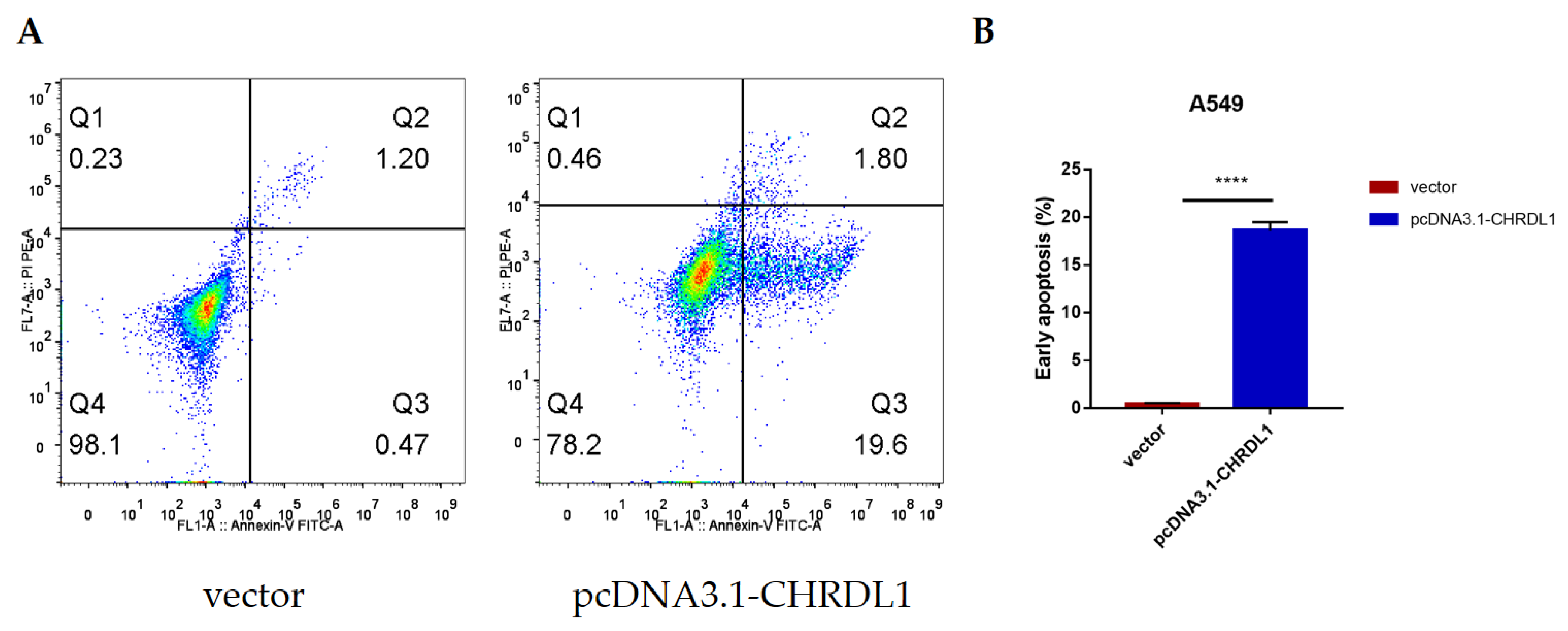
3.6. CHRDL1 Could Accelerate the Early Apoptosis of Lung Adenocarcinoma Cell Line A549
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Chalela, R.; Curull, V.; Enriquez, C.; Pijuan, L.; Bellosillo, B.; Gea, J. Lung adenocarcinoma: From molecular basis to genome-guided therapy and immunotherapy. J. Thorac. Dis. 2017, 9, 2142–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saintigny, P.; Burger, J.A. Recent advances in non-small cell lung cancer biology and clinical management. Discov. Med. 2012, 13, 287–297. [Google Scholar]
- Calvayrac, O.; Pradines, A.; Pons, E.; Mazieres, J.; Guibert, N. Molecular biomarkers for lung adenocarcinoma. Eur Respir. J. 2017, 49, 1601734. [Google Scholar] [CrossRef]
- Yoneda, K.; Imanishi, N.; Ichiki, Y.; Tanaka, F. Treatment of Non-small Cell Lung Cancer with EGFR-mutations. J. UOEH 2019, 41, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.J.; Shaw, A.T. Recent Advances in Targeting ROS1 in Lung Cancer. J. Thorac. Oncol. 2017, 12, 1611–1625. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Jiang, T.; Fang, Z.; Tang, S.; Cheng, R.; Li, Y.; Ren, S.; Su, C.; Min, W.; Guo, X.; Zhu, W.; et al. Mutational Landscape and Evolutionary Pattern of Liver and Brain Metastasis in Lung Adenocarcinoma. J. Thorac. Oncol. 2021, 16, 237–249. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, D.; Zhang, Y.; Ding, Z.; Zheng, Y.; Chen, S.; Wan, Y. Integrative analysis of mRNA and miRNA expression profiles reveals seven potential diagnostic biomarkers for nonsmall cell lung cancer. Oncol. Rep. 2020, 43, 99–112. [Google Scholar] [PubMed]
- Ghafouri-Fard, S.; Shoorei, H.; Branicki, W.; Taheri, M. Non-coding RNA profile in lung cancer. Exp. Mol. Pathol. 2020, 114, 104411. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, S.; Debernardi, A.; Jacquiau, B.; Vitte, A.L.; Vesin, A.; Nagy-Mignotte, H.; Moro-Sibilot, D.; Brichon, P.Y.; Lantuejoul, S.; Hainaut, P.; et al. Ectopic activation of germline and placental genes identifies aggressive metastasis-prone lung cancers. Sci. Transl. Med. 2013, 5, 186ra66. [Google Scholar] [CrossRef] [Green Version]
- Botling, J.; Edlund, K.; Lohr, M.; Hellwig, B.; Holmberg, L.; Lambe, M.; Berglund, A.; Ekman, S.; Bergqvist, M.; Ponten, F.; et al. Biomarker discovery in non-small cell lung cancer: Integrating gene expression profiling, meta-analysis, and tissue microarray validation. Clin. Cancer Res. 2013, 19, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Der, S.D.; Sykes, J.; Pintilie, M.; Zhu, C.Q.; Strumpf, D.; Liu, N.; Jurisica, I.; Shepherd, F.A.; Tsao, M.S. Validation of a histology-independent prognostic gene signature for early-stage, non-small-cell lung cancer including stage IA patients. J. Thorac. Oncol. 2014, 9, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Fertig, E.J.; Jaffe, A.E.; Storey, J.D.; Zhang, Y.; Torres, L.C. sva: Surrogate Variable Analysis; R Package Version 3.36.0. 2020. Available online: http://bioconductor.org/packages/3.14/bioc/html/sva.html (accessed on 14 April 2021).
- Wilkerson, M.D.; Yin, X.; Walter, V.; Zhao, N.; Cabanski, C.R.; Hayward, M.C.; Miller, C.R.; Socinski, M.A.; Parsons, A.M.; Thorne, L.B.; et al. Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PLoS ONE 2012, 7, e36530. [Google Scholar] [CrossRef] [Green Version]
- Okayama, H.; Kohno, T.; Ishii, Y.; Shimada, Y.; Shiraishi, K.; Iwakawa, R.; Furuta, K.; Tsuta, K.; Shibata, T.; Yamamoto, S.; et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012, 72, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Schabath, M.B.; Welsh, E.A.; Fulp, W.J.; Chen, L.; Teer, J.K.; Thompson, Z.J.; Engel, B.E.; Xie, M.; Berglund, A.E.; Creelan, B.C.; et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene 2016, 35, 3209–3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinforma. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant Graphics for Data Analysis, 2nd edition. Meas.-Interdiscip. Res. Perspect. 2019, 17, 160–167. [Google Scholar] [CrossRef]
- Therneau, T. A Package for Survival Analysis in R. R Package Version 3.2-7. 2021. Available online: https://cran.r-project.org/web/packages/survival/vignettes/survival.pdf (accessed on 12 May 2021).
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Blanche, P.; Dartigues, J.F.; Jacqmin-Gadda, H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 2013, 32, 5381–5397. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Gordon, M.; Lumley, T. forestplot: Advanced Forest Plot Using ‘grid’ Graphics. R Package Version 1.10. 2020. Available online: https://CRAN.R-project.org/package=forestplot (accessed on 12 May 2021).
- Harrell, F.E. rms: Regression Modeling Strategies. R Package Version 6.2-0. 2021. Available online: https://CRAN.R-project.org/package=survival (accessed on 12 May 2021).
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Carlson, M.R.; Zhang, B.; Fang, Z.; Mischel, P.S.; Horvath, S.; Nelson, S.F. Gene connectivity, function, and sequence conservation: Predictions from modular yeast co-expression networks. BMC Genomics 2006, 7, 40. [Google Scholar] [CrossRef]
- Yang, L.; Xu, Y.; Yan, Y.; Luo, P.; Chen, S.; Zheng, B.; Yan, W.; Chen, Y.; Wang, C. Common Nevus and Skin Cutaneous Melanoma: Prognostic Genes Identified by Gene Co-Expression Network Analysis. Genes 2019, 10, 747. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Pei, F.; Yang, F.; Li, L.; Amin, A.D.; Liu, S.; Buchan, J.R.; Cho, W.C. Role of Autophagy and Apoptosis in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2017, 18, 367. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xuan, Y.; Gao, B.; Sun, X.; Miao, S.; Lu, T.; Wang, Y.; Jiao, W. Identification of an eight-gene prognostic signature for lung adenocarcinoma. Cancer Manag. Res. 2018, 10, 3383–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, Z.; Yu, Z. Identification of a novel glycolysis-related gene signature for predicting metastasis and survival in patients with lung adenocarcinoma. J. Transl. Med. 2019, 17, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wu, L.; Ao, H.; Zhao, M.; Leng, X.; Liu, M.; Ma, J.; Zhu, J. Prognostic implications of autophagy-associated gene signatures in non-small cell lung cancer. Aging 2019, 11, 11440–11462. [Google Scholar] [CrossRef]
- Zuo, S.; Wei, M.; Zhang, H.; Chen, A.; Wu, J.; Wei, J.; Dong, J. A robust six-gene prognostic signature for prediction of both disease-free and overall survival in non-small cell lung cancer. J. Transl. Med. 2019, 17, 152. [Google Scholar] [CrossRef]
- Ferrell, J.M.; Chiang, J.Y. Circadian rhythms in liver metabolism and disease. Acta Pharm. Sin. B 2015, 5, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Reszka, E.; Zienolddiny, S. Epigenetic Basis of Circadian Rhythm Disruption in Cancer. Methods Mol. Biol. 2018, 1856, 173–201. [Google Scholar]
- Hitzler, J.K.; Soares, H.D.; Drolet, D.W.; Inaba, T.; O’Connel, S.; Rosenfeld, M.G.; Morgan, J.I.; Look, A.T. Expression patterns of the hepatic leukemia factor gene in the nervous system of developing and adult mice. Brain Res. 1999, 820, 1–11. [Google Scholar] [CrossRef]
- Suzuki, K.; Yoshida, K.; Ueha, T.; Kaneshiro, K.; Nakai, A.; Hashimoto, N.; Uchida, K.; Hashimoto, T.; Kawasaki, Y.; Shibanuma, N.; et al. Methotrexate upregulates circadian transcriptional factors PAR bZIP to induce apoptosis on rheumatoid arthritis synovial fibroblasts. Arthritis Res. Ther. 2018, 20, 55. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, Y.; Ni, C.; Meng, G.; Sheng, X. HLF/miR-132/TTK axis regulates cell proliferation, metastasis and radiosensitivity of glioma cells. Biomed. Pharmacother. 2016, 83, 898–904. [Google Scholar] [CrossRef]
- Wahlestedt, M.; Ladopoulos, V.; Hidalgo, I.; Castillo, M.S.; Hannah, R.; Sawen, P.; Wan, H.; Dudenhoffer-Pfeifer, M.; Magnusson, M.; Norddahl, G.L.; et al. Critical Modulation of Hematopoietic Lineage Fate by Hepatic Leukemia Factor. Cell Rep. 2017, 21, 2251–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, O.; Beraza, N. Hepatocellular carcinomas: Evolution to sorafenib resistance through hepatic leukaemia factor. Gut 2019, 68, 1728–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Liu, A.; Lin, Z.; Wang, B.; Chai, X.; Chen, S.; Lu, W.; Zheng, M.; Cao, T.; Zhong, M.; et al. Downregulation of the circadian rhythm regulator HLF promotes multiple-organ distant metastases in non-small cell lung cancer through PPAR/NF-κb signaling. Cancer Lett. 2020, 482, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Troilo, H.; Barrett, A.L.; Wohl, A.P.; Jowitt, T.A.; Collins, R.F.; Bayley, C.P.; Zuk, A.V.; Sengle, G.; Baldock, C. The role of chordin fragments generated by partial tolloid cleavage in regulating BMP activity. Biochem. Soc Trans. 2015, 43, 795–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawala, A.; Sutcliffe, C.; Ashe, H.L. Multistep molecular mechanism for bone morphogenetic protein extracellular transport in the Drosophila embryo. Proc. Natl. Acad. Sci. USA 2012, 109, 11222–11227. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Nagai, A.; Sheikh, A.M.; Mitaki, S.; Wakabayashi, K.; Kim, S.U.; Kobayashi, S.; Yamaguchi, S. A human neural stem cell line provides neuroprotection and improves neurological performance by early intervention of neuroinflammatory system. Brain Res. 2016, 1631, 194–203. [Google Scholar] [CrossRef]
- Pei, Y.F.; Zhang, Y.J.; Lei, Y.; Wu, W.D.; Ma, T.H.; Liu, X.Q. Hypermethylation of the CHRDL1 promoter induces proliferation and metastasis by activating Akt and Erk in gastric cancer. Oncotarget 2017, 8, 23155–23166. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yang, L.; Zhong, T.; Mueller, M.; Men, Y.; Zhang, N.; Xie, J.; Giang, K.; Chung, H.; Sun, X.; et al. H19 lncRNA alters DNA methylation genome wide by regulating S-adenosylhomocysteine hydrolase. Nat. Commun. 2015, 6, 10221. [Google Scholar] [CrossRef] [Green Version]
- Cyr-Depauw, C.; Northey, J.J.; Tabaries, S.; Annis, M.G.; Dong, Z.; Cory, S.; Hallett, M.; Rennhack, J.P.; Andrechek, E.R.; Siegel, P.M. Chordin-Like 1 Suppresses Bone Morphogenetic Protein 4-Induced Breast Cancer Cell Migration and Invasion. Mol. Cell. Biol. 2016, 36, 1509–1525. [Google Scholar] [CrossRef] [Green Version]
- Pohl, N.M.; Tong, C.; Fang, W.; Bi, X.; Li, T.; Yang, W. Transcriptional regulation and biological functions of selenium-binding protein 1 in colorectal cancer in vitro and in nude mouse xenografts. PLoS ONE 2009, 4, e7774. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wang, H.; Miller, C.T.; Thomas, D.G.; Gharib, T.G.; Misek, D.E.; Giordano, T.J.; Orringer, M.B.; Hanash, S.M.; Beer, D.G. Reduced selenium-binding protein 1 expression is associated with poor outcome in lung adenocarcinomas. J. Pathol. 2004, 202, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Raucci, R.; Colonna, G.; Guerriero, E.; Capone, F.; Accardo, M.; Castello, G.; Costantini, S. Structural and functional studies of the human selenium binding protein-1 and its involvement in hepatocellular carcinoma. Biochim. Biophys. Acta 2011, 1814, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kang, H.J.; You, K.T.; Kim, S.H.; Lee, K.Y.; Kim, T.I.; Kim, C.; Song, S.Y.; Kim, H.J.; Lee, C.; et al. Suppression of human selenium-binding protein 1 is a late event in colorectal carcinogenesis and is associated with poor survival. Proteomics 2006, 6, 3466–3476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, F.; Younes, M.; Liu, H.; Chen, C.; Yao, Q. Reduced Selenium-Binding Protein 1 in Breast Cancer Correlates with Poor Survival and Resistance to the Anti-Proliferative Effects of Selenium. PLoS ONE 2013, 8, e63702. [Google Scholar] [CrossRef] [Green Version]
- Caswell, D.R.; Chuang, C.H.; Ma, R.K.; Winters, I.P.; Snyder, E.L.; Winslow, M.M. Tumor Suppressor Activity of Selenbp1, a Direct Nkx2-1 Target, in Lung Adenocarcinoma. Mol. Cancer Res. 2018, 16, 1737–1749. [Google Scholar] [CrossRef] [Green Version]
- Burre, J.; Zimmermann, H.; Volknandt, W. Identification and characterization of SV31, a novel synaptic vesicle membrane protein and potential transporter. J. Neurochem. 2007, 103, 276–287. [Google Scholar] [CrossRef]
- Sanchez, V.B.; Ali, S.; Escobar, A.; Cuajungco, M.P. Transmembrane 163 (TMEM163) protein effluxes zinc. Arch. Biochem. Biophys. 2019, 677, 108166. [Google Scholar] [CrossRef]
- Cuajungco, M.P.; Basilio, L.C.; Silva, J.; Hart, T.; Tringali, J.; Chen, C.C.; Biel, M.; Grimm, C. Cellular zinc levels are modulated by TRPML1-TMEM163 interaction. Traffic 2014, 15, 1247–1265. [Google Scholar] [CrossRef] [Green Version]
- Cuajungco, M.P.; Kiselyov, K. The mucolipin-1 (TRPML1) ion channel, transmembrane-163 (TMEM163) protein, and lysosomal zinc handling. Front. Biosci. (Landmark Ed.) 2017, 22, 1330–1343. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Training Dataset | Validation Dataset | |||||||
|---|---|---|---|---|---|---|---|---|---|
| GSE26939 | p Value $ | GSE31210 | p Value $ | GSE72094 | p Value $ | TCGA-LUAD | p Value $ | ||
| Number of patients | 321 | 116 | 226 | 442 | 500 | ||||
| Platforms | GPL570 | GPL9053 | GPL570 | GPL15048 | GDC Data Portal | ||||
| Gender | |||||||||
| Male | 179 | 49 | 0.2518 | 105 | 0.0370 * | 240 | 0.7128 | 230 | 0.0066 ** |
| Female | 142 | 51 | 121 | 202 | 270 | ||||
| Unknown | 0 | 16 | 0 | 0 | 0 | ||||
| Age (years) | 64.90 ± 9.92 | 64.04 ± 10.88 | 0.4359 | 59.58 ± 7.40 | <0.0001 **** | 69.30 ± 9.33 | <0.0001 **** | 65.26 ± 10.05 | 0.6244 |
| Clinical Stage | |||||||||
| Unknown | 0 | 29 | 0 | 28 | 8 | ||||
| Stage I | 234 | 55 | <0.0001 **** | 168 | 0.0050 ** | 265 | <0.0001 **** | 268 | <0.0001 **** |
| Stage II | 70 | 14 | 58 | 69 | 119 | ||||
| Stage III | 13 | 16 | 0 | 63 | 80 | ||||
| Stage IV | 4 | 2 | 0 | 17 | 25 | ||||
| Relapse | |||||||||
| No | 174 | Null | 162 | 0.1728 | Null | 289 | 0.0360 * | ||
| Yes | 91 | 64 | 211 | ||||||
| Unknown | 56 | 20 | 0 | ||||||
| Follow up | |||||||||
| Alive | 145 | 49 | 0.6629 | 191 | <0.0001 **** | 298 | <0.0001 **** | 318 | <0.0001 **** |
| Dead | 176 | 66 | 35 | 122 | 182 | ||||
| Unknown | 0 | 1 | 20 | 22 | 0 | ||||
| Affy ID | Ensembl ID | Gene Symbol | Gene Description | Regression Coefficient | R | p Value |
|---|---|---|---|---|---|---|
| 204753_s_at | ENSG00000108924 | HLF | hepatic leukemia factor | −0.03400109 | 0.756241276 | 3.33 × 10−62 |
| 209763_at | ENSG00000101938 | CHRDL1 | chordin-like 1 | −0.06167218 | 0.702915849 | 2.58 × 10−50 |
| 214433_s_at | ENSG00000143416 | SELENBP1 | selenium binding protein 1 | −0.16551196 | 0.771245541 | 4.04 × 10−66 |
| 223503_at | ENSG00000152128 | TMEM163 | transmembrane protein 163 | −0.01203028 | 0.703663266 | 1.84 × 10−50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Yang, L.; Zhang, L.; Zheng, X.; Xu, H.; Wang, K.; Weng, X. Identification of a Four-Gene Signature Associated with the Prognosis Prediction of Lung Adenocarcinoma Based on Integrated Bioinformatics Analysis. Genes 2022, 13, 238. https://doi.org/10.3390/genes13020238
Wu Y, Yang L, Zhang L, Zheng X, Xu H, Wang K, Weng X. Identification of a Four-Gene Signature Associated with the Prognosis Prediction of Lung Adenocarcinoma Based on Integrated Bioinformatics Analysis. Genes. 2022; 13(2):238. https://doi.org/10.3390/genes13020238
Chicago/Turabian StyleWu, Yuan, Lingge Yang, Long Zhang, Xinjie Zheng, Huan Xu, Kai Wang, and Xianwu Weng. 2022. "Identification of a Four-Gene Signature Associated with the Prognosis Prediction of Lung Adenocarcinoma Based on Integrated Bioinformatics Analysis" Genes 13, no. 2: 238. https://doi.org/10.3390/genes13020238