
Differential Allele-Specific Expression Revealed Functional Variants and Candidate Genes Related to Meat Quality Traits in B. indicus Muscle
 ,
,  , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Production and Sample Collection
2.2. Phenotype Measurements
2.3. DNA Extraction, Genotyping and Whole Genome Sequencing
2.4. RNA Extraction and Sequencing
2.5. Analysis of Differential Allele-Specific Expression (DASE) between Contrasting Groups for Meat Quality Phenotypes
2.6. Functional Annotation and Gene Enrichment Analyses
2.7. Co-Expression Analysis
2.8. Data Integration
3. Results
3.1. The Muscle-Related Phenotypes Are Correlated
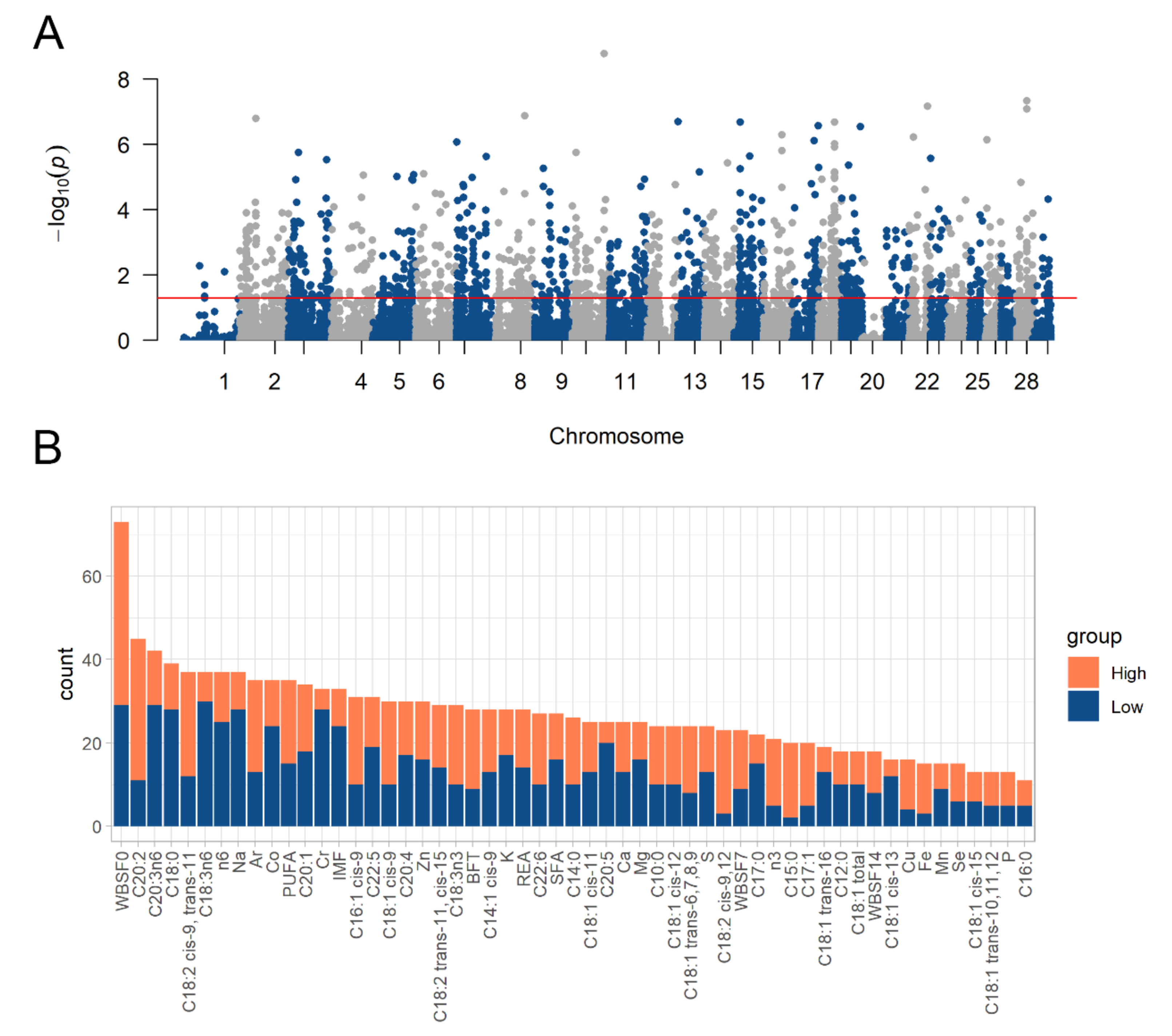
3.2. Differential Allelic Imbalance Related to Livestock Traits Was Identified in Bovine Muscle
3.3. Genes with Differential Allele-Specific Expression Were Related to Muscle Homeostasis
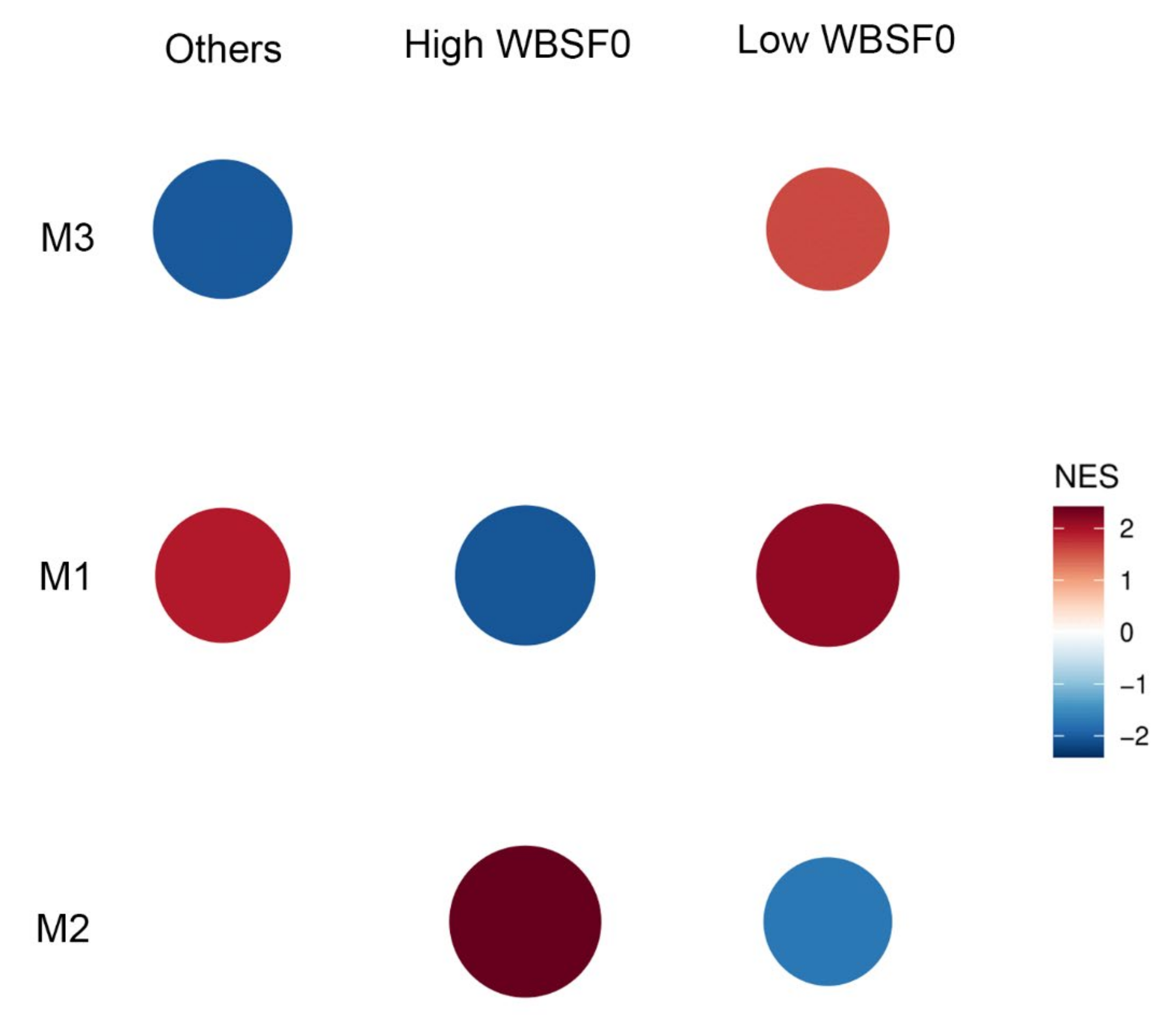
3.4. Co-Expression between DASE Genes
3.5. Integration Was Found between DASE SNPs and QTLs for the Related Phenotypes
3.6. Regulatory Variants Related to the DASE Pattern
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castel, S.E.; Levy-Moonshine, A.; Mohammadi, P.; Banks, E.; Lappalainen, T. Tools and Best Practices for Data Processing in Allelic Expression Analysis. Genome Biol. 2015, 16, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Hormozdiari, F.; Jew, B.; Castel, S.E.; Lappalainen, T.; Ernst, J.; Sul, J.H.; Eskin, E. Leveraging Allelic Imbalance to Refine Fine-Mapping for EQTL Studies. PLoS Genet. 2019, 15, e1008481. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, K.; Tilghman, S.M. Allele-Specific Gene Expression in Mammals: The Curious Case of the Imprinted RNAs. Genes Dev. 1994, 8, 1867–1874. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, A.J.; Vander Jagt, C.J.; Hayes, B.J.; Khansefid, M.; Marett, L.C.; Millen, C.A.; Nguyen, T.T.T.; Goddard, M.E. Extensive Variation between Tissues in Allele Specific Expression in an Outbred Mammal. BMC Genom. 2015, 16, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.-C.; Ferris, E.; Cheng, T.; Hörndli, C.S.; Gleason, K.; Tamminga, C.; Wagner, J.D.; Boucher, K.M.; Christian, J.L.; Gregg, C. Diverse Non-Genetic, Allele-Specific Expression Effects Shape Genetic Architecture at the Cellular Level in the Mammalian Brain. Neuron 2017, 93, 1094–1109.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khansefid, M.; Pryce, J.E.; Bolormaa, S.; Chen, Y.; Millen, C.A.; Chamberlain, A.J.; Vander Jagt, C.J.; Goddard, M.E. Comparing Allele Specific Expression and Local Expression Quantitative Trait Loci and the Influence of Gene Expression on Complex Trait Variation in Cattle. BMC Genom. 2018, 19, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Devarajan, K.; Zhou, Y.; Slater, C.M.; Daly, M.B.; Chen, X. Identifying Breast Cancer Risk Loci by Global Differential Allele-Specific Expression (DASE) Analysis in Mammary Epithelial Transcriptome. BMC Genom. 2012, 13, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, M.M.; Zerlotini, A.; Rocha, M.I.P.; Bruscadin, J.J.; Diniz, W.J.d.S.; Cardoso, T.F.; Cesar, A.S.M.; Afonso, J.; Andrade, B.G.N.; Mudadu, M.d.A.; et al. Allele-Specific Expression Is Widespread in Bos Indicus Muscle and Affects Meat Quality Candidate Genes. Sci. Rep. 2020, 10, 10204. [Google Scholar] [CrossRef]
- Bruscadin, J.J.; Cardoso, T.F.; da Silva Diniz, W.J.; Afonso, J.; de Souza, M.M.; Petrini, J.; de Andrade, B.G.; da Silva, V.H.; Ferraz, J.B.S.; Zerlotini, A.; et al. Allele-Specific Expression Reveals Functional SNPs Affecting Muscle-Related Genes in Bovine. Biochim. Biophys. Acta (BBA) 2022, 194886. [Google Scholar] [CrossRef]
- Guillocheau, G.M.; El Hou, A.; Meersseman, C.; Esquerré, D.; Rebours, E.; Letaief, R.; Simao, M.; Hypolite, N.; Bourneuf, E.; Bruneau, N.; et al. Survey of Allele Specific Expression in Bovine Muscle. Sci. Rep. 2019, 9, 4297. [Google Scholar] [CrossRef] [PubMed]
- Varkoohi, S.; Banabazi, M.H.; Ghsemi-Siab, M. Allele Specific Expression (Ase) Analysis between Bos Taurus and Bos Indicus Cows Using Rna-Seq Data at Snp Level and Gene Level. An. Acad. Bras. DeCienc. 2021, 93, 1–9. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, P.A. Transcriptomics in the RNA-Seq Era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Przytycki, P.F.; Singh, M. Differential Allele-Specific Expression Uncovers Breast Cancer Genes Dysregulated by Cis Noncoding Mutations. Cell Syst. 2020, 10, 193–203.e4. [Google Scholar] [CrossRef] [PubMed]
- Tizioto, P.C.; Meirelles, S.L.; Veneroni, G.B.; Tullio, R.R.; Rosa, A.N.; Alencar, M.M.; Medeiros, S.R.; Siqueira, F.; Feijó, G.L.D.; Silva, L.O.C.; et al. A SNP in ASAP1 Gene Is Associated with Meat Quality and Production Traits in Nelore Breed. Meat Sci. 2012, 92, 855–857. [Google Scholar] [CrossRef]
- Tizioto, P.C.; Decker, J.E.; Taylor, J.F.; Schnabel, R.D.; Mudadu, M.A.; Silva, F.L.; Mourao, G.B.; Coutinho, L.L.; Tholon, P.; Sonstegard, T.S.; et al. Genome Scan for Meat Quality Traits in Nelore Beef Cattle. Physiol. Genom. 2013, 45, 1012–1020. [Google Scholar] [CrossRef] [Green Version]
- Cesar, A.S.; Regitano, L.C.; Mourão, G.B.; Tullio, R.R.; Lanna, D.P.; Nassu, R.T.; Mudado, M.A.; Oliveira, P.S.; do Nascimento, M.L.; Chaves, A.S.; et al. Genome-Wide Association Study for Intramuscular Fat Deposition and Composition in Nellore Cattle. BMC Genet. 2014, 15, 39. [Google Scholar] [CrossRef] [Green Version]
- Tizioto, P.C.; Gromboni, C.F.; de Araujo Nogueira, A.R.; de Souza, M.M.; de Alvarenga Mudadu, M.; Tholon, P.; Rosa, A.d.N.; Tullio, R.R.; Medeiros, S.R.; Nassu, R.T.; et al. Calcium and Potassium Content in Beef: Influences on Tenderness and Associations with Molecular Markers in Nellore Cattle. Meat Sci. 2014, 96, 436–440. [Google Scholar] [CrossRef]
- Gonçalves, T.M.; de Almeida Regitano, L.C.; Koltes, J.E.; Cesar, A.S.M.; da Silva Andrade, S.C.; Mourão, G.B.; Gasparin, G.; Moreira, G.C.M.; Fritz-Waters, E.; Reecy, J.M.; et al. Gene Co-Expression Analysis Indicates Potential Pathways and Regulators of Beef Tenderness in Nellore Cattle. Front. Genet. 2018, 9, 441. [Google Scholar] [CrossRef] [Green Version]
- Cesar, A.S.M.; Regitano, L.C.A.; Poleti, M.D.; Andrade, S.C.S.; Tizioto, P.C.; Oliveira, P.S.N.; Felício, A.M.; do Nascimento, M.L.; Chaves, A.S.; Lanna, D.P.D.; et al. Differences in the Skeletal Muscle Transcriptome Profile Associated with Extreme Values of Fatty Acids Content. BMC Genom. 2016, 17, 961. [Google Scholar] [CrossRef] [Green Version]
- Diniz, W.J.d.S.; Coutinho, L.L.; Tizioto, P.C.; Cesar, A.S.M.; Gromboni, C.F.; Nogueira, A.R.A.; de Oliveira, P.S.N.; Souza, M.M.d.; Regitano, L.C.d.A. Iron Content Affects Lipogenic Gene Expression in the Muscle of Nelore Beef Cattle. PLoS ONE 2016, 11, e0161160. [Google Scholar] [CrossRef]
- Silva-Vignato, B.; Coutinho, L.L.; Cesar, A.S.M.; Poleti, M.D.; Regitano, L.C.A.; Balieiro, J.C.C. Comparative Muscle Transcriptome Associated with Carcass Traits of Nellore Cattle. BMC Genom. 2017, 18, 506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonso, J.; Coutinho, L.L.; Tizioto, P.C.; da Silva Diniz, W.J.; de Lima, A.O.; Rocha, M.I.P.; Buss, C.E.; Andrade, B.G.N.; Piaya, O.; da Silva, J.V.; et al. Muscle Transcriptome Analysis Reveals Genes and Metabolic Pathways Related to Mineral Concentration in Bos Indicus. Sci. Rep. 2019, 9, 12715. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, T.F.; Coutinho, L.L.; Bruscadin, J.J.; da Silva Diniz, W.J.; Petrini, J.; Andrade, B.G.N.; de Oliveira, P.S.N.; Poleti, M.D.; Cesar, A.S.M.; da Silveira, J.C.; et al. Multi-Omics Approach Reveals MiR-SNPs Affecting Muscle Fatty Acids Profile in Nelore Cattle. Genes 2021, 12, 67. [Google Scholar] [CrossRef]
- Mudadu, M.A.; Porto-Neto, L.R.; Mokry, F.B.; Tizioto, P.C.; Oliveira, P.S.N.; Tullio, R.R.; Nassu, R.T.; Niciura, S.C.M.; Tholon, P.; Alencar, M.M.; et al. Genomic Structure and Marker-Derived Gene Networks for Growth and Meat Quality Traits of Brazilian Nelore Beef Cattle. BMC Genom. 2016, 17, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhbannikov, I.Y.; Hunter, S.S.; Foster, J.A.; Settles, M.L. SeqyClean: A Pipeline for High-Throughput Sequence Data Preprocessing. In Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Boston, MA, USA, 20 August 2017; pp. 407–416. [Google Scholar]
- Cesar, A.S.M.; Regitano, L.C.A.; Reecy, J.M.; Poleti, M.D.; Oliveira, P.S.N.; de Oliveira, G.B.; Moreira, G.C.M.; Mudadu, M.A.; Tizioto, P.C.; Koltes, J.E.; et al. Identification of Putative Regulatory Regions and Transcription Factors Associated with Intramuscular Fat Content Traits. BMC Genom. 2018, 19, 499. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Van De Geijn, B.; Mcvicker, G.; Gilad, Y.; Pritchard, J.K. WASP: Allele-Specific Software for Robust Molecular Quantitative Trait Locus Discovery. Nat. Methods 2015, 12, 1061–1063. [Google Scholar] [CrossRef] [Green Version]
- Degner, J.F.; Marioni, J.C.; Pai, A.A.; Pickrell, J.K.; Nkadori, E.; Gilad, Y.; Pritchard, J.K. Effect of Read-Mapping Biases on Detecting Allele-Specific Expression from RNA-Sequencing Data. Bioinformatics 2009, 25, 3207–3212. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, P.S.T.; Ferreira, G.R.; Cardozo, L.E.; Bürger, M.C.; Arias-Carrasco, R.; Maruyama, S.R.; Hirata, T.D.C.; Lima, D.S.; Passos, F.M.; Fukutani, K.F.; et al. CEMiTool: A Bioconductor Package for Performing Comprehensive Modular Co-Expression Analyses. BMC Bioinform. 2018, 19, 56. [Google Scholar] [CrossRef] [Green Version]
- Tizioto, P.C.; Taylor, J.F.; Decker, J.E.; Gromboni, C.F.; Mudadu, M.A.; Schnabel, R.D.; Coutinho, L.L.; Mourão, G.B.; Oliveira, P.S.; Souza, M.M.; et al. Detection of Quantitative Trait Loci for Mineral Content of Nelore Longissimus Dorsi Muscle. Genet. Sel. Evol. 2015, 47, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, M.M.; Niciura, S.C.M.; Rocha, M.I.P.; Pan, Z.; Zhou, H.; Bruscadin, J.J.; da Silva Diniz, W.J.; Afonso, J.; de Oliveira, P.S.N.; Mourão, G.B.; et al. DNA Methylation May Affect Beef Tenderness through Signal Transduction in Bos Indicus. Epigenetics Chromatin 2022, 15, 15. [Google Scholar] [CrossRef]
- Kern, C.; Wang, Y.; Xu, X.; Pan, Z.; Halstead, M.; Chanthavixay, G.; Saelao, P.; Waters, S.; Xiang, R.; Chamberlain, A.; et al. Functional Annotations of Three Domestic Animal Genomes Provide Vital Resources for Comparative and Agricultural Research. Nat. Commun. 2021, 12, 1821. [Google Scholar] [CrossRef]
- Gel, B.; Díez-Villanueva, A.; Serra, E.; Buschbeck, M.; Peinado, M.A.; Malinverni, R. RegioneR: An R/Bioconductor Package for the Association Analysis of Genomic Regions Based on Permutation Tests. Bioinformatics 2015, btv562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dronkelaar, C.; van Velzen, A.; Abdelrazek, M.; van der Steen, A.; Weijs, P.J.M.; Tieland, M. Minerals and Sarcopenia; The Role of Calcium, Iron, Magnesium, Phosphorus, Potassium, Selenium, Sodium, and Zinc on Muscle Mass, Muscle Strength, and Physical Performance in Older Adults: A Systematic Review. J. Am. Med Dir. Assoc. 2018, 19, 6–11.e3. [Google Scholar] [CrossRef]
- Cunha, T.A.; Vermeulen-Serpa, K.M.; Grilo, E.C.; Leite-Lais, L.; Brandão-Neto, J.; Vale, S.H.L. Association between Zinc and Body Composition: An Integrative Review. J. Trace Elem. Med. Biol. 2022, 71, 126940. [Google Scholar] [CrossRef] [PubMed]
- Ghassemi Nejad, J.; Lee, B.-H.; Kim, B.-W.; Ohh, S.-J.; Sung, K.I. Effects of Chromium Methionine Supplementation on Blood Metabolites and Fatty Acid Profile of Beef during Late Fattening Period in Holstein Steers. Asian-Australas. J. Anim. Sci. 2016, 29, 378–383. [Google Scholar] [CrossRef]
- Garmyn, A.J.; Hilton, G.G.; Mateescu, R.G.; Morgan, J.B.; Reecy, J.M.; Tait, R.G.; Beitz, D.C.; Duan, Q.; Schoonmaker, J.P.; Mayes, M.S.; et al. Estimation of Relationships between Mineral Concentration and Fatty Acid Composition of Longissimus Muscle and Beef Palatability Traits1. J. Anim. Sci. 2011, 89, 2849–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Listrat, A.; Gagaoua, M.; Andueza, D.; Gruffat, D.; Normand, J.; Mairesse, G.; Picard, B.; Hocquette, J.-F. What Are the Drivers of Beef Sensory Quality Using Metadata of Intramuscular Connective Tissue, Fatty Acids and Muscle Fiber Characteristics? Livest. Sci. 2020, 240, 104209. [Google Scholar] [CrossRef]
- Mukherjee, R.; Das, A.; Chakrabarti, S.; Chakrabarti, O. Calcium Dependent Regulation of Protein Ubiquitination–Interplay between E3 Ligases and Calcium Binding Proteins. BBA-Mol. Cell Res. 2017, 1864, 1227–1235. [Google Scholar] [CrossRef]
- Ando, H.; Watabe, H.; Valencia, J.C.; Yasumoto, K.; Furumura, M.; Funasaka, Y.; Oka, M.; Ichihashi, M.; Hearing, V.J. Fatty Acids Regulate Pigmentation via Proteasomal Degradation of Tyrosinase. J. Biol. Chem. 2004, 279, 15427–15433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supakankul, P.; Mekchay, S. Effect of UBE3C Polymorphisms on Intramuscular Fat Content and Fatty Acid Composition in Duroc Pigs. Genet. Mol. Res. 2016, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Kemp, C.M.; Sensky, P.L.; Bardsley, R.G.; Buttery, P.J.; Parr, T. Tenderness – An Enzymatic View. Meat Sci. 2010, 84, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Furuno, T.; Kanno, T.; Arita, K.; Asami, M.; Utsumi, T.; Doi, Y.; Inoue, M.; Utsumi, K. Roles of Long Chain Fatty Acids and Carnitine in Mitochondrial Membrane Permeability Transition11Abbreviations: CsA, Cyclosporin A; Cyt. c, Cytochrome c; FBS, Fetal Bovine Serum; Pal-CoA, Palmitoyl-CoA; Pi, Inorganic Phosphate; LCFA, Long Chain Fatty Acid; MPT, Membrane Permeability Transition; PLA2, Phospholipase A2; RCR, Respiratory Control Ratio; and T3, 3,3′,5-Triiodothyronine. Biochem. Pharmacol. 2001, 62, 1037–1046. [Google Scholar] [CrossRef]
- Matarneh, S.K.; England, E.M.; Scheffler, T.L.; Gerrard, D.E. The Conversion of Muscle to Meat. In Lawrie’s Meat Science; Elsevier: Amsterdam, The Netherlands, 2017; pp. 159–185. ISBN 978-0-08-100694-8. [Google Scholar]
- Letts, J.A.; Sazanov, L.A. Clarifying the Supercomplex: The Higher-Order Organization of the Mitochondrial Electron Transport Chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Zurek, M.P.; Turner, A.A.; Khobeir, A.; Yazdi, N.; Hakim, L.; Liu, J.; Vaishnav, A.; et al. Cytochrome c Phosphorylation: Control of Mitochondrial Electron Transport Chain Flux and Apoptosis. Int. J. Biochem. Cell Biol. 2020, 121, 105704. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-L.; Yu, Q.-L.; Han, L.; Ma, X.-L.; Song, R.-D.; Zhao, S.-N.; Zhang, W.-H. Study on the Effect of Reactive Oxygen Species-Mediated Oxidative Stress on the Activation of Mitochondrial Apoptosis and the Tenderness of Yak Meat. Food Chem. 2018, 244, 394–402. [Google Scholar] [CrossRef]
- Liu, C.; Wei, Q.; Li, X.; Han, D.; Liu, J.; Huang, F.; Zhang, C. Proteomic Analyses of Mitochondrial Damage in Postmortem Beef Muscles. J. Sci. Food Agric. 2022, jsfa.11767. [Google Scholar] [CrossRef] [PubMed]
- Albesa, M.; Ogrodnik, J.; Rougier, J.-S.; Abriel, H. Regulation of the Cardiac Sodium Channel Nav1.5 by Utrophin in Dystrophin-Deficient Mice. Cardiovasc. Res. 2011, 89, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, T.L.; Moir, L.; Hemming, S.; Edwards, B.; Squire, S.; Davies, K.; Guiraud, S. Utrophin Influences Mitochondrial Pathology and Oxidative Stress in Dystrophic Muscle. Skelet. Muscle 2017, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Sartori, R.; Schirwis, E.; Blaauw, B.; Bortolanza, S.; Zhao, J.; Enzo, E.; Stantzou, A.; Mouisel, E.; Toniolo, L.; Ferry, A.; et al. BMP Signaling Controls Muscle Mass. Nat. Genet. 2013, 45, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Zhang, L.; Wang, W.; Wu, S. Effect of Genetic Variants in the SMAD1 and SMAD5 Genes Promoter on Growth and Beef Quality Traits in Cattle. Gene 2022, 819, 146220. [Google Scholar] [CrossRef]
- Zhang, R.; Miao, J.; Song, Y.; Zhang, W.; Xu, L.; Chen, Y.; Zhang, L.; Gao, H.; Zhu, B.; Li, J.; et al. Genome-Wide Association Study Identifies the PLAG1-OXR1 Region on BTA14 for Carcass Meat Yield in Cattle. Physiol. Genom. 2019, 51, 137–144. [Google Scholar] [CrossRef]
- Wu, Y.; Davies, K.E.; Oliver, P.L. The Antioxidant Protein Oxr1 Influences Aspects of Mitochondrial Morphology. Free Radic. Biol. Med. 2016, 95, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Durand, M.; Kolpak, A.; Farrell, T.; Elliott, N.A.; Shao, W.; Brown, M.; Volkert, M.R. The OXR Domain Defines a Conserved Family of Eukaryotic Oxidation Resistance Proteins. BMC Cell Biol. 2007, 8, 13. [Google Scholar] [CrossRef]
- Xu, B.; Wang, H.; Wright, S.; Hyle, J.; Zhang, Y.; Shao, Y.; Niu, M.; Fan, Y.; Rosikiewicz, W.; Djekidel, M.N.; et al. Acute Depletion of CTCF Rewires Genome-Wide Chromatin Accessibility. Genome Biol. 2021, 22, 244. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DASE SNP | Trait (DASE Analysis) | Trait (QTL Integration) |
|---|---|---|
| rs720456892, rs43664623, chr11:63468724 G>A, chr11:69714613 G>A | C18:0 | C18:0 |
| rs716062365, rs41721088, rs109947761 | REA | REA |
| rs109976566, rs444293703 | WBSF7 | WBSF7 |
| rs208417619, rs720858445 | WBSF0 | WBSF7 |
| DASE SNP | Variant Annotation/ Gene | Trait (DASE Analysis) | Trait (DE) | FAANG | Trait (QTL) | aseQTL | cis-eQTL |
|---|---|---|---|---|---|---|---|
| rs457578905 | synonymous_variant/THBS4 | Cr | P, Cu, Na, K, Mg, Average daily gain | ATAC-seq | LFAT | No | no |
| rs718541482 | 3_prime_UTR_variant/WDR48 | PUFA | CLA-c9t11, PA | ATAC-seq | REA | No | no |
| rs211442363 | 3_prime_UTR_variant/SPARC | C20:1 | IMF | H3K27ac | REA | No | no |
| rs715527852 | downstream_gene_variant/SUGT1 | P, S | CLA-c9t11 | ATAC-seq | REA | No | no |
| rs1115255230 | synonymous_variant/SPARC | n6, C20:1, C10:0 | IMF | H3K27ac | REA | No | no |
| rs209388096 | synonymous_variant/AMFR | C22:6 | REA | ATAC-seq | no | Yes | no |
| rs719946630 | synonymous_variant/PGD | WBSF14 | CLA-c9t11 | ATAC-seq | no | Yes | no |
| rs1117381943 | upstream_gene_variant/CAVIN2 | C18_1_T6_T7_T8_T9 | AO | H3K4me3, H3K27ac, H3K4me1, H3K4me3, ATAC-seq, CTCF | no | Yes | no |
| rs516592412 | synonymous_variant/PDG | C20:1 | CLA-c9t11 | ATAC-seq | no | Yes | no |
| rs1117068355 | upstream_gene_variant/CAVIN2 | C18_1_T6_T7_T8_T9 | AO | H3K4me3, H3K4me1, H3K27me3, H3K27ac, ATAC-seq, CTCF | no | Yes | no |
| rs109090536 | 5_prime_UTR_variant/DCAF11 | C20:2 | AO | H3K4me3, H3K4me1, H3K27ac | no | No | yes |
| rs443738741 | synonymous_variant/LNX2 | C20:2 | REA | ATAC-seq | no | No | yes |
| rs379719524 | synonymous_variant/NRAP | Cr | AO | ATAC-seq | no | No | yes |
| rs715652252 | synonymous_variant/LNX2 | C20:2 | REA | ATAC-seq, CTCF | no | No | yes |
| rs468833876 | upstream_gene_variant/NUTF2 | C18_2_C9_C12 | CLA-c9t11 | H3K27ac, H3K27me3, H3K4me3 | no | No | yes |
| rs135451771 | synonymous_variant/NRAP | Cr | AO | ATAC-seq | no | No | yes |
| chr2:5447956 G>A | synonymous_variant/BIN1 | C18_1_T6_T7_T8_T9 | AO | H3K27ac | no | No | yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruscadin, J.J.; Cardoso, T.F.; da Silva Diniz, W.J.; de Souza, M.M.; Afonso, J.; Vieira, D.; Malheiros, J.; Andrade, B.G.N.; Petrini, J.; Ferraz, J.B.S.; et al. Differential Allele-Specific Expression Revealed Functional Variants and Candidate Genes Related to Meat Quality Traits in B. indicus Muscle. Genes 2022, 13, 2336. https://doi.org/10.3390/genes13122336
Bruscadin JJ, Cardoso TF, da Silva Diniz WJ, de Souza MM, Afonso J, Vieira D, Malheiros J, Andrade BGN, Petrini J, Ferraz JBS, et al. Differential Allele-Specific Expression Revealed Functional Variants and Candidate Genes Related to Meat Quality Traits in B. indicus Muscle. Genes. 2022; 13(12):2336. https://doi.org/10.3390/genes13122336
Chicago/Turabian StyleBruscadin, Jennifer Jessica, Tainã Figueiredo Cardoso, Wellison Jarles da Silva Diniz, Marcela Maria de Souza, Juliana Afonso, Dielson Vieira, Jessica Malheiros, Bruno Gabriel Nascimento Andrade, Juliana Petrini, José Bento Sterman Ferraz, and et al. 2022. "Differential Allele-Specific Expression Revealed Functional Variants and Candidate Genes Related to Meat Quality Traits in B. indicus Muscle" Genes 13, no. 12: 2336. https://doi.org/10.3390/genes13122336