
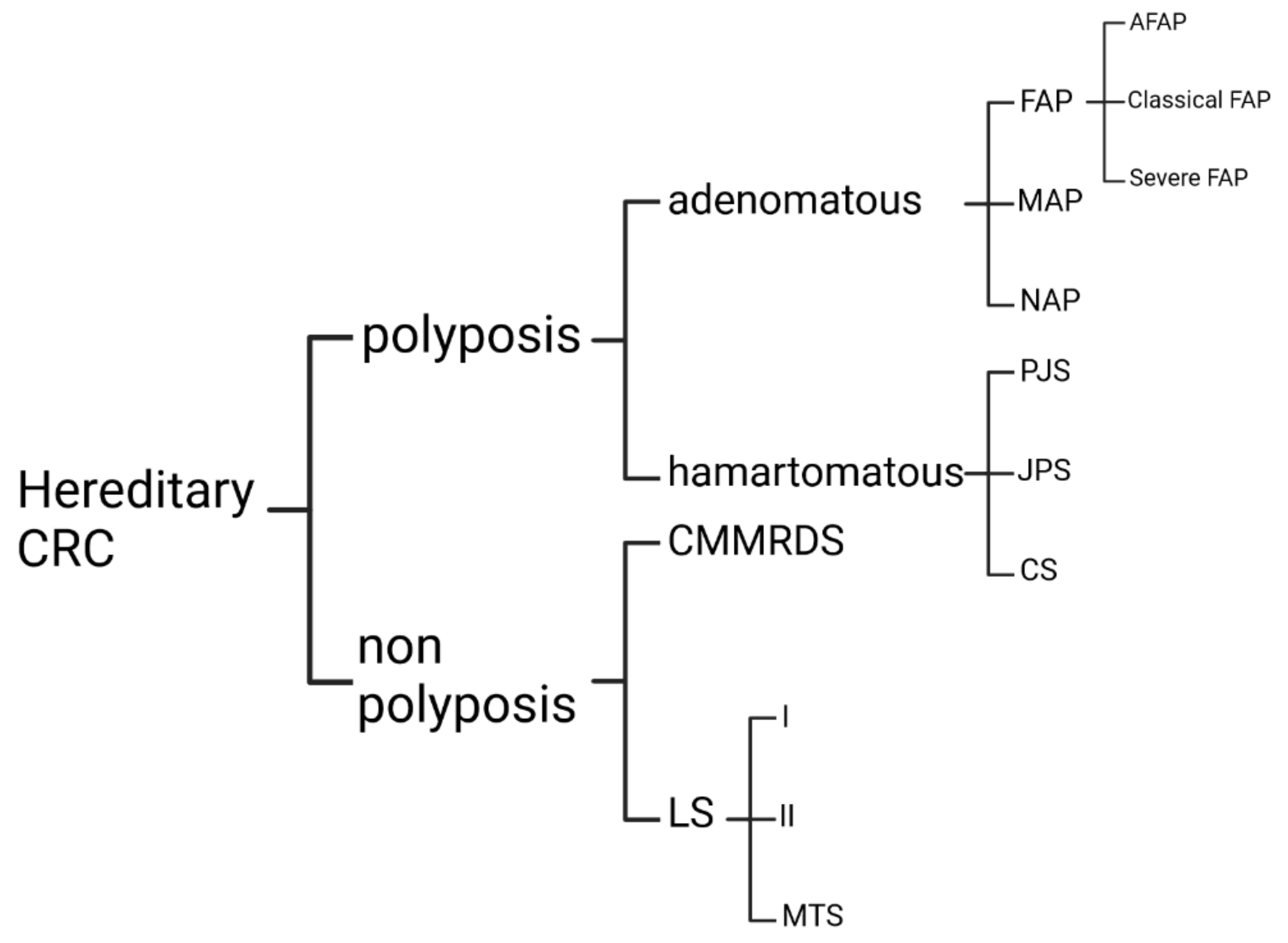
Strong Hereditary Predispositions to Colorectal Cancer
 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Nonpolyposis CRC Predisposition
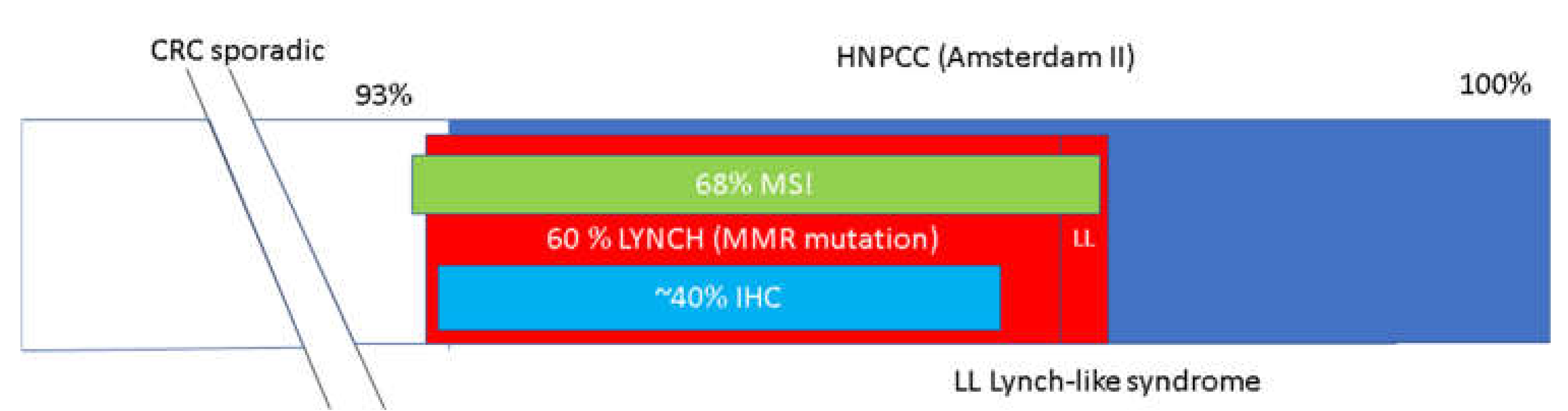
3.1.1. Lynch Syndrome
3.1.2. Constitutional Mismatch Repair Deficiency Syndrome
3.2. Adenomatous Polyps
3.2.1. Familial Adenomatous Polyposis
3.2.2. FAP Classification
3.3. Hamartomatous Polyposis
3.3.1. Peutz–Jeghers Syndrome
3.3.2. Juvenile Polyposis Syndrome
3.3.3. Cowden Syndrome
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The Ever-increasing Importance of Cancer as a Leading Cause of Premature Death Worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef]
- All Cancers Fact Sheet. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/39-All-cancers-fact-sheet.pdf (accessed on 12 October 2022).
- Weiderpass, E.; Stewart, B.W. World Cancer Report: Cancer Research for Cancer Prevention; IARC Press: Lyon, France, 2020; ISBN 978-92-832-0447-3. [Google Scholar]
- Fearon, E.R.; Vogelstein, B. A Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Stoffel, E.M.; Kastrinos, F. Familial Colorectal Cancer, Beyond Lynch Syndrome. Clin. Gastroenterol. Hepatol. 2014, 12, 1059–1068. [Google Scholar] [CrossRef] [Green Version]
- Knudson, A.G. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Vasen, H.F.A.; Mecklin, J.-P.; Meera Khan, P.; Lynch, H.T. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis. Colon Rectum 1991, 34, 424–425. [Google Scholar] [CrossRef]
- Vasen, H.; Watson, P.; Mecklin, J.; Lynch, H. New Clinical Criteria for Hereditary Nonpolyposis Colorectal Cancer (HNPCC, Lynch Syndrome) Proposed by the International Collaborative Group on HNPCC☆. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef]
- Rodriguez-Bigas, M.A.; Boland, C.R.; Hamilton, S.R.; Henson, D.E.; Srivastava, S.; Jass, J.R.; Khan, P.M.; Lynch, H.; Smyrk, T.; Perucho, M.; et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: Meeting Highlights and Bethesda Guidelines. JNCI J. Natl. Cancer Inst. 1997, 89, 1758–1762. [Google Scholar] [CrossRef] [Green Version]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Ruschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. JNCI J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Sammour, T.; Hayes, I.P.; Hill, A.G.; Macrae, F.A.; Winter, D.C. Familial Colorectal Cancer Syndromes: An Overview of Clinical Management. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 757–764. [Google Scholar] [CrossRef]
- Lynch, H.T.; de la Chapelle, A. Hereditary Colorectal Cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef]
- Lorans, M.; Dow, E.; Macrae, F.A.; Winship, I.M.; Buchanan, D.D. Update on Hereditary Colorectal Cancer: Improving the Clinical Utility of Multigene Panel Testing. Clin. Color. Cancer 2018, 17, e293–e305. [Google Scholar] [CrossRef] [Green Version]
- Valle, L. Recent Discoveries in the Genetics of Familial Colorectal Cancer and Polyposis. Clin. Gastroenterol. Hepatol. 2017, 15, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, K.; Kratz, C.P.; Vasen, H.F.A.; Caron, O.; Colas, C.; Entz-Werle, N.; Gerdes, A.-M.; Goldberg, Y.; Ilencikova, D.; Muleris, M.; et al. Diagnostic Criteria for Constitutional Mismatch Repair Deficiency Syndrome: Suggestions of the European Consortium “care for CMMRD” (C4CMMRD). J. Med. Genet. 2014, 51, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Grady, W.M.; Carethers, J.M. Genomic and Epigenetic Instability in Colorectal Cancer Pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef] [Green Version]
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [Green Version]
- Boland, C.R.; Goel, A. Microsatellite Instability in Colorectal Cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef]
- Hsieh, P.; Yamane, K. DNA Mismatch Repair: Molecular Mechanism, Cancer, and Ageing. Mech. Ageing Dev. 2008, 129, 391–407. [Google Scholar] [CrossRef] [Green Version]
- Stojic, L.; Mojas, N.; Cejka, P.; di Pietro, M.; Ferrari, S.; Marra, G.; Jiricny, J. Mismatch Repair-Dependent G2 Checkpoint Induced by Low Doses of SN1 Type Methylating Agents Requires the ATR Kinase. Genes Dev. 2004, 18, 1331–1344. [Google Scholar] [CrossRef] [Green Version]
- Lao, V.V.; Grady, W.M. Epigenetics and Colorectal Cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef] [Green Version]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG Island Methylator Phenotype Underlies Sporadic Microsatellite Instability and Is Tightly Associated with BRAF Mutation in Colorectal Cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Furukawa, Y.; Nakamura, Y.; Matsubara, N.; Ishikawa, H.; Arai, M.; Tomita, N.; Tamura, K.; Sugano, K.; Ishioka, C.; et al. Comparison of Clinical Features between Suspected Familial Colorectal Cancer Type X and Lynch Syndrome in Japanese Patients with Colorectal Cancer: A Cross-Sectional Study Conducted by the Japanese Society for Cancer of the Colon and Rectum. Jpn. J. Clin. Oncol. 2015, 45, 153–159. [Google Scholar] [CrossRef]
- Carethers, J.M.; Stoffel, E.M. Lynch Syndrome and Lynch Syndrome Mimics: The Growing Complex Landscape of Hereditary Colon Cancer. World J. Gastroenterol. 2015, 21, 9253–9261. [Google Scholar] [CrossRef]
- Win, A.K.; Jenkins, M.A.; Buchanan, D.D.; Clendenning, M.; Young, J.P.; Giles, G.G.; Goldblatt, J.; Leggett, B.A.; Hopper, J.L.; Thibodeau, S.N.; et al. Determining the Frequency of de Novo Germline Mutations in DNA Mismatch Repair Genes. J. Med. Genet. 2011, 48, 530–534. [Google Scholar] [CrossRef]
- Xavier, A.; Olsen, M.F.; Lavik, L.A.; Johansen, J.; Singh, A.K.; Sjursen, W.; Scott, R.J.; Talseth-Palmer, B.A. Comprehensive Mismatch Repair Gene Panel Identifies Variants in Patients with Lynch-like Syndrome. Mol. Genet. Genom. Med. 2019, 7, e850. [Google Scholar] [CrossRef] [Green Version]
- Lynch, H.T.; Smyrk, T.C.; Watson, P.; Lanspa, S.J.; Lynch, J.F.; Lynch, P.M.; Cavalieri, R.J.; Boland, C.R. Genetics, Natural History, Tumor Spectrum, and Pathology of Hereditary Nonpolyposis Colorectal Cancer: An Updated Review. Gastroenterology 1993, 104, 1535–1549. [Google Scholar] [CrossRef]
- Muir, E.G.; Bell, A.J.Y.; Barlow, K.A. Multiple Primary Carcinomata of the Colon, Duodenum, and Larynx Associated with Kerato-Acanthomata of the Face. Br. J. Surg. 1967, 54, 191–195. [Google Scholar] [CrossRef]
- Torre, D. Society Transactions: New York Dermatological Society. Arch. Dermatol. 1967, 1968, 549–551. [Google Scholar]
- Balmaña, J.; Balaguer, F.; Cervantes, A.; Arnold, D. Familial Risk-Colorectal Cancer: ESMO Clinical Practice Guidelines. Ann. Oncol. 2013, 24, vi73–vi80. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Goel, A.; Hornick, J.L.; Sen, A.; Turgeon, D.K.; Ruffin, M.T.; Marcon, N.E.; Baron, J.A.; Bresalier, R.S.; Syngal, S.; et al. Microsatellite Instability and DNA Mismatch Repair Protein Deficiency in Lynch Syndrome Colorectal Polyps. Cancer Prev. Res. 2012, 5, 574–582. [Google Scholar] [CrossRef] [Green Version]
- Willett, C.G.; Chang, D.T.; Czito, B.G.; Meyer, J.; Wo, J. The Cancer Genome Atlas Network Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Huang, J.; Papadopoulos, N.; McKinley, A.J.; Farrington, S.M.; Curtis, L.J.; Wyllie, A.H.; Zheng, S.; Willson, J.K.; Markowitz, S.D.; Morin, P.; et al. APC Mutations in Colorectal Tumors with Mismatch Repair Deficiency. Proc. Natl. Acad. Sci. USA 1996, 93, 9049–9054. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the Type II TGF-β Receptor in Colon Cancer Cells with Microsatellite Instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef]
- Rampino, N.; Yamamoto, H.; Ionov, Y.; Li, Y.; Sawai, H.; Reed, J.C.; Perucho, M. Somatic Frameshift Mutations in the BAX Gene in Colon Cancers of the Microsatellite Mutator Phenotype. Science 1997, 275, 967–969. [Google Scholar] [CrossRef]
- Ahadova, A.; Gallon, R.; Gebert, J.; Ballhausen, A.; Endris, V.; Kirchner, M.; Stenzinger, A.; Burn, J.; von Knebel Doeberitz, M.; Bläker, H.; et al. Three Molecular Pathways Model Colorectal Carcinogenesis in Lynch Syndrome: Three Pathways of CRC in Lynch Syndrome. Int. J. Cancer 2018, 143, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Kloor, M.; Huth, C.; Voigt, A.Y.; Benner, A.; Schirmacher, P.; von Knebel Doeberitz, M.; Bläker, H. Prevalence of Mismatch Repair-Deficient Crypt Foci in Lynch Syndrome: A Pathological Study. Lancet Oncol. 2012, 13, 598–606. [Google Scholar] [CrossRef]
- Ahadova, A.; von Knebel Doeberitz, M.; Bläker, H.; Kloor, M. CTNNB1-Mutant Colorectal Carcinomas with Immediate Invasive Growth: A Model of Interval Cancers in Lynch Syndrome. Fam. Cancer 2016, 15, 579–586. [Google Scholar] [CrossRef]
- Møller, P.; Seppälä, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Incidence of and Survival after Subsequent Cancers in Carriers of Pathogenic MMR Variants with Previous Cancer: A Report from the Prospective Lynch Syndrome Database. Gut 2017, 66, 1657–1664. [Google Scholar] [CrossRef] [Green Version]
- Warthin, A.S. Heredity with Reference to Carcinoma as Shown by the Study of the Cases Examined in the Pathological Laboratory of the University of Michigan, 1895–1913. CA A Cancer J. Clin. 1985, 35, 348–359. [Google Scholar] [CrossRef]
- Lynch, H.T.; Shaw, M.W.; Magnuson, C.W.; Larsen, A.L.; Krush, A.J. Hereditary Factors in Cancer. Study of Two Large Midwestern Kindreds. Arch. Intern. Med. 1966, 117, 206–212. [Google Scholar] [CrossRef]
- Lynch, H.T.; Lynch, P.M.; Pester, J.; Fusaro, R.M. The Cancer Family Syndrome. Rare Cutaneous Phenotypic Linkage of Torre’s Syndrome. Arch. Intern. Med. 1981, 141, 607–611. [Google Scholar] [CrossRef]
- Peltomäki, P.; Lothe, R.A.; Aaltonen, L.A.; Pylkkänen, L.; Nyström-Lahti, M.; Seruca, R.; David, L.; Holm, R.; Ryberg, D.; Haugen, A. Microsatellite Instability Is Associated with Tumors That Characterize the Hereditary Non-Polyposis Colorectal Carcinoma Syndrome. Cancer Res. 1993, 53, 5853–5855. [Google Scholar]
- Lindblom, A.; Tannergård, P.; Werelius, B.; Nordenskjöld, M. Genetic Mapping of a Second Locus Predisposing to Hereditary Non–Polyposis Colon Cancer. Nat. Genet. 1993, 5, 279–282. [Google Scholar] [CrossRef]
- Plazzer, J.P.; Sijmons, R.H.; Woods, M.O.; Peltomäki, P.; Thompson, B.; Den Dunnen, J.T.; Macrae, F. The InSiGHT Database: Utilizing 100 Years of Insights into Lynch Syndrome. Fam. Cancer 2013, 12, 175–180. [Google Scholar] [CrossRef]
- Nyström-Lahti, M.; Kristo, P.; Nicolaides, N.C.; Chang, S.-Y.; Aaltonen, L.A.; Moisio, A.-L.; Järvinen, H.J.; Mecklin, J.-P.; Kinzler, K.W.; Vogelstein, B.; et al. Founding Mutations and Alu-Mediated Recombination in Hereditary Colon Cancer. Nat. Med. 1995, 1, 1203–1206. [Google Scholar] [CrossRef]
- Wijnen, J.; van der Klift, H.; Vasen, H.; Khan, P.M.; Menko, F.; Tops, C.; Meijers Heijboer, H.; Lindhout, D.; Møller, P.; Fodde, R. MSH2 Genomic Deletions Are a Frequent Cause of HNPCC. Nat. Genet. 1998, 20, 326–328. [Google Scholar] [CrossRef]
- Engel, C.; Ahadova, A.; Seppälä, T.T.; Aretz, S.; Bigirwamungu-Bargeman, M.; Bläker, H.; Bucksch, K.; Büttner, R.; de Vos Tot Nederveen Cappel, W.T.; Endris, V.; et al. Associations of Pathogenic Variants in MLH1, MSH2, and MSH6 With Risk of Colorectal Adenomas and Tumors and With Somatic Mutations in Patients with Lynch Syndrome. Gastroenterology 2020, 158, 1326–1333. [Google Scholar] [CrossRef]
- Mangold, E. A Genotype-Phenotype Correlation in HNPCC: Strong Predominance of Msh2 Mutations in 41 Patients with Muir-Torre Syndrome. J. Med. Genet. 2004, 41, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A. Atypical HNPCC Owing to MSH6 Germline Mutations: Analysis of a Large Dutch Pedigree. J. Med. Genet. 2001, 38, 318–322. [Google Scholar] [CrossRef] [Green Version]
- Berends, M.J.W.; Wu, Y.; Sijmons, R.H.; Mensink, R.G.J.; van der Sluis, T.; Hordijk-Hos, J.M.; de Vries, E.G.E.; Hollema, H.; Karrenbeld, A.; Buys, C.H.C.M.; et al. Molecular and Clinical Characteristics of MSH6 Variants: An Analysis of 25 Index Carriers of a Germline Variant. Am. J. Hum. Genet. 2002, 70, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Verma, L.; Kane, M.F.; Brassett, C.; Schmeits, J.; Evans, D.G.; Kolodner, R.D.; Maher, E.R. Mononucleotide Microsatellite Instability and Germline MSH6 Mutation Analysis in Early Onset Colorectal Cancer. J. Med. Genet. 1999, 36, 678–682. [Google Scholar]
- Senter, L.; Clendenning, M.; Sotamaa, K.; Hampel, H.; Green, J.; Potter, J.D.; Lindblom, A.; Lagerstedt, K.; Thibodeau, S.N.; Lindor, N.M.; et al. The Clinical Phenotype of Lynch Syndrome Due to Germ-Line PMS2 Mutations. Gastroenterology 2008, 135, 419–428.e1. [Google Scholar] [CrossRef] [Green Version]
- Engel, C.; Rahner, N.; Schulmann, K.; Holinski–Feder, E.; Goecke, T.O.; Schackert, H.K.; Kloor, M.; Steinke, V.; Vogelsang, H.; Möslein, G.; et al. Efficacy of Annual Colonoscopic Surveillance in Individuals with Hereditary Nonpolyposis Colorectal Cancer. Clin. Gastroenterol. Hepatol. 2010, 8, 174–182. [Google Scholar] [CrossRef]
- Järvinen, H.J.; Aarnio, M.; Mustonen, H.; Aktan–Collan, K.; Aaltonen, L.A.; Peltomäki, P.; Chapelle, A.D.L.; Mecklin, J. Controlled 15-Year Trial on Screening for Colorectal Cancer in Families with Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 2000, 118, 829–834. [Google Scholar] [CrossRef]
- Ahadova, A.; Seppälä, T.T.; Engel, C.; Gallon, R.; Burn, J.; Holinski-Feder, E.; Steinke-Lange, V.; Möslein, G.; Nielsen, M.; Ten Broeke, S.W.; et al. The “Unnatural” History of Colorectal Cancer in Lynch Syndrome: Lessons from Colonoscopy Surveillance. Int. J. Cancer 2021, 148, 800–811. [Google Scholar] [CrossRef]
- Møller, P.; Seppälä, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Cancer Incidence and Survival in Lynch Syndrome Patients Receiving Colonoscopic and Gynaecological Surveillance: First Report from the Prospective Lynch Syndrome Database. Gut 2017, 66, 464–472. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E.; Boland, C.R.; Burke, C.A.; Burt, R.W.; Church, J.M.; Dominitz, J.A.; Johnson, D.A.; Kaltenbach, T.; et al. Guidelines on Genetic Evaluation and Management of Lynch Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2014, 147, 502–526. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Wang, S.; Pan, P.; Xia, T.; Chang, X.; Yang, X.; Guo, L.; Meng, Q.; Yang, F.; Qian, W.; et al. Magnitude, Risk Factors, and Factors Associated with Adenoma Miss Rate of Tandem Colonoscopy: A Systematic Review and Meta-Analysis. Gastroenterology 2019, 156, 1661–1674.e11. [Google Scholar] [CrossRef]
- Stryker, S.J.; Wolff, B.G.; Culp, C.E.; Libbe, S.D.; Ilstrup, D.M.; MacCarty, R.L. Natural History of Untreated Colonic Polyps. Gastroenterology 1987, 93, 1009–1013. [Google Scholar] [CrossRef]
- Seppälä, T.; Pylvänäinen, K.; Evans, D.G.; Järvinen, H.; Renkonen-Sinisalo, L.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Lindblom, A.; Macrae, F.; et al. Colorectal Cancer Incidence in Path_MLH1 Carriers Subjected to Different Follow-up Protocols: A Prospective Lynch Syndrome Database Report. Hered. Cancer Clin. Pract. 2017, 15, 18. [Google Scholar] [CrossRef] [Green Version]
- Pai, R.K.; Dudley, B.; Karloski, E.; Brand, R.E.; O’Callaghan, N.; Rosty, C.; Buchanan, D.D.; Jenkins, M.A.; Thibodeau, S.N.; French, A.J.; et al. DNA Mismatch Repair Protein Deficient Non-Neoplastic Colonic Crypts: A Novel Indicator of Lynch Syndrome. Mod. Pathol. 2018, 31, 1608–1618. [Google Scholar] [CrossRef]
- Seppälä, T.T.; Ahadova, A.; Dominguez-Valentin, M.; Macrae, F.; Evans, D.G.; Therkildsen, C.; Sampson, J.; Scott, R.; Burn, J.; Möslein, G.; et al. Lack of Association between Screening Interval and Cancer Stage in Lynch Syndrome May Be Accounted for by Over-Diagnosis; a Prospective Lynch Syndrome Database Report. Hered. Cancer Clin. Pract. 2019, 17, 8. [Google Scholar] [CrossRef]
- Linnebacher, M.; Gebert, J.; Rudy, W.; Woerner, S.; Yuan, Y.P.; Bork, P.; von Knebel Doeberitz, M. Frameshift Peptide-Derived T-Cell Epitopes: A Source of Novel Tumor-Specific Antigens. Int. J. Cancer 2001, 93, 6–11. [Google Scholar] [CrossRef]
- Drago, L.; Toscano, M.; De Grandi, R.; Casini, V.; Pace, F. Persisting Changes of Intestinal Microbiota after Bowel Lavage and Colonoscopy. Eur. J. Gastroenterol. Hepatol. 2016, 28, 532–537. [Google Scholar] [CrossRef]
- Backes, Y.; Seerden, T.C.J.; van Gestel, R.S.F.E.; Kranenburg, O.; Ubink, I.; Schiffelers, R.M.; van Straten, D.; van der Capellen, M.S.; van de Weerd, S.; de Leng, W.W.J.; et al. Tumor Seeding During Colonoscopy as a Possible Cause for Metachronous Colorectal Cancer. Gastroenterology 2019, 157, 1222–1232.e4. [Google Scholar] [CrossRef] [Green Version]
- Burn, J.; Gerdes, A.-M.; Macrae, F.; Mecklin, J.-P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Long-Term Effect of Aspirin on Cancer Risk in Carriers of Hereditary Colorectal Cancer: An Analysis from the CAPP2 Randomised Controlled Trial. Lancet 2011, 378, 2081–2087. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 276300. Available online: https://omim.org/ (accessed on 19 May 2021).
- Abedalthagafi, M. Constitutional Mismatch Repair-Deficiency: Current Problems and Emerging Therapeutic Strategies. Oncotarget 2018, 9, 35458–35469. [Google Scholar] [CrossRef] [Green Version]
- Suerink, M.; Potjer, T.P.; Versluijs, A.B.; ten Broeke, S.W.; Tops, C.M.; Wimmer, K.; Nielsen, M. Constitutional Mismatch Repair Deficiency in a Healthy Child: On the Spot Diagnosis? Clin. Genet. 2018, 93, 134–137. [Google Scholar] [CrossRef] [Green Version]
- Felton, K.E.A.; Gilchrist, D.M.; Andrew, S.E. Constitutive Deficiency in DNA Mismatch Repair: Is It Time for Lynch III? Clin. Genet. 2007, 71, 499–500. [Google Scholar] [CrossRef]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting from Germline Biallelic Mismatch Repair Deficiency. JCO 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Gilligan, B.M.; Yuan, J.; Li, T. Current Status and Perspectives in Translational Biomarker Research for PD-1/PD-L1 Immune Checkpoint Blockade Therapy. J. Hematol. Oncol. 2016, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Bussey, H.J.R. Familial Polyposis Coli: Family Studies, Histopathology, Differential Diagnosis, and Results of Treatment; Johns Hopkins University Press: Baltimore, MD, USA, 1975; ISBN 978-0-8018-1686-4. [Google Scholar]
- Sarre, R.G.; Frost, A.G.; Jagelman, D.G.; Petras, R.E.; Sivak, M.V.; McGannon, E. Gastric and Duodenal Polyps in Familial Adenomatous Polyposis: A Prospective Study of the Nature and Prevalence of Upper Gastrointestinal Polyps. Gut 1987, 28, 306–314. [Google Scholar] [CrossRef]
- Petersen, G.M.; Slack, J.; Nakamura, Y. Screening Guidelines and Premorbid Diagnosis of Familial Adenomatous Polyposis Using Linkage. Gastroenterology 1991, 100, 1658–1664. [Google Scholar] [CrossRef]
- Distante, S.; Nasioulas, S.; Somers, G.R.; Cameron, D.J.; Young, M.A.; Forrest, S.M.; Gardner, R.J. Familial Adenomatous Polyposis in a 5 Year Old Child: A Clinical, Pathological, and Molecular Genetic Study. J. Med. Genet. 1996, 33, 157–160. [Google Scholar] [CrossRef]
- Plawski, A.; Podralska, M.; Krokowicz, P.; Paszkowski, J.; Lubiński, J.; Słomski, R. Rodzinna polipowatość gruczolakowata jelita grubego. Postępy Nauk Med. 2008, 7, 553–561. [Google Scholar]
- Bisgaard, M.L.; Fenger, K.; Bülow, S.; Niebuhr, E.; Mohr, J. Familial Adenomatous Polyposis (FAP): Frequency, Penetrance, and Mutation Rate. Hum. Mutat. 1994, 3, 121–125. [Google Scholar] [CrossRef]
- Groden, J.; Thliveris, A.; Samowitz, W.; Carlson, M.; Gelbert, L.; Albertsen, H.; Joslyn, G.; Stevens, J.; Spirio, L.; Robertson, M.; et al. Identification and Characterization of the Familial Adenomatous Polyposis Coli Gene. Cell 1991, 66, 589–600. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Clendenning, M.; Sotamaa, K.; Prior, T.; Westman, J.A.; et al. Feasibility of Screening for Lynch Syndrome Among Patients with Colorectal Cancer. JCO 2008, 26, 5783–5788. [Google Scholar] [CrossRef] [Green Version]
- Spigelman, A.D.; Talbot, I.C.; Williams, C.B.; Domizio, P.; Phillips, R.K.S. Upper gastrointestinal cancer in patients with familial adenomatous polyposis. Lancet 1989, 334, 783–785. [Google Scholar] [CrossRef]
- Plail, R.O.; Bussey, H.J.R.; Glazer, G.; Thomson, J.P.S. Adenomatous Polyposis: An Association with Carcinoma of the Thyroid. Br. J. Surg. 1987, 74, 377–380. [Google Scholar] [CrossRef]
- Krawczuk-Rybak, M.; Jakubiuk-Tomaszuk, A.; Skiba, E.; Plawski, A. Hepatoblastoma as a Result of APC Gene Mutation. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 334–336. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Offerhaus, G.J.; Lee, D.H.; Krush, A.J.; Tersmette, A.C.; Booker, S.V.; Kelley, N.C.; Hamilton, S.R. Increased Risk of Thyroid and Pancreatic Carcinoma in Familial Adenomatous Polyposis. Gut 1993, 34, 1394–1396. [Google Scholar] [CrossRef] [Green Version]
- Kropilak, M.; Jagelman, D.G.; Fazio, V.W.; Lavery, I.L.; McGannon, E. Brain Tumors in Familial Adenomatous Polyposis. Dis. Colon Rectum 1989, 32, 778–782. [Google Scholar] [CrossRef]
- Johnson Smith, T.G.P.; Clark, S.K.; Katz, D.E.; Reznek, R.H.; Phillips, R.K.S. Adrenal Masses Are Associated with Familial Adenomatous Polyposis. Dis. Colon Rectum 2000, 43, 1739–1742. [Google Scholar] [CrossRef]
- Gardner, E.J. A Genetic and Clinical Study of Intestinal Polyposis, a Predisposing Factor for Carcinoma of the Colon and Rectum. Am. J. Hum. Genet. 1951, 3, 167–176. [Google Scholar]
- Kubo, K.; Miyatani, H.; Takenoshita, Y.; Abe, K.; Oka, M.; Iida, M.; Itoh, H. Widespread Radiopacity of Jaw Bones in Familial Adenomatosis Coli. J. Cranio-Maxillofac. Surg. 1989, 17, 350–353. [Google Scholar] [CrossRef]
- Traboulsi, E.I.; Krush, A.J.; Gardner, E.J.; Booker, S.V.; Offerhaus, G.J.A.; Yardley, J.H.; Hamilton, S.R.; Luk, G.D.; Giardiello, F.M.; Welsh, S.B.; et al. Prevalence and Importance of Pigmented Ocular Fundus Lesions in Gardner’s Syndrome. N. Engl. J. Med. 1987, 316, 661–667. [Google Scholar] [CrossRef]
- Wallis, Y.L.; Macdonald, F.; Morton, J.E.V.; McKeown, C.M.; Neoptolemos, J.; Keighley, M.; Morton, D.G. Genotype-Phenotype Correlation between Position of Constitutional APC Gene Mutation and CHRPE Expression in Familial Adenomatous Polyposis. Hum. Genet. 1994, 94, 543–548. [Google Scholar] [CrossRef]
- Gurbuz, A.K.; Giardiello, F.M.; Petersen, G.M.; Krush, A.J.; Offerhaus, G.J.; Booker, S.V.; Kerr, M.C.; Hamilton, S.R. Desmoid Tumours in Familial Adenomatous Polyposis. Gut 1994, 35, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Nugent, K.P.; Spigelman, A.D.; Phillips, R.K.S. Life Expectancy after Colectomy and Ileorectal Anastomosis for Familial Adenomatous Polyposis. Dis. Colon Rectum 1993, 36, 1059–1062. [Google Scholar] [CrossRef]
- Half, E.; Bercovich, D.; Rozen, P. Familial Adenomatous Polyposis. Orphanet. J. Rare Dis. 2009, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Winawer, S.J.; Fletcher, R.H.; Miller, L.; Godlee, F.; Stolar, M.H.; Mulrow, C.D.; Woolf, S.H.; Glick, S.N.; Ganiats, T.G.; Bond, J.H.; et al. Colorectal Cancer Screening: Clinical Guidelines and Rationale. Gastroenterology 1997, 112, 594–642. [Google Scholar] [CrossRef]
- Burt, R.W.; Cannon, J.A.; David, D.S.; Early, D.S.; Ford, J.M.; Giardiello, F.M.; Halverson, A.L.; Hamilton, S.R.; Hampel, H.; Ismail, M.K.; et al. Colorectal Cancer Screening. J. Natl. Compr. Canc. Netw. 2013, 11, 1538–1575. [Google Scholar] [CrossRef] [Green Version]
- Bodmer, W.F.; Bailey, C.J.; Bodmer, J.; Bussey, H.J.R.; Ellis, A.; Gorman, P.; Lucibello, F.C.; Murday, V.A.; Rider, S.H.; Scambler, P.; et al. Localization of the Gene for Familial Adenomatous Polyposis on Chromosome 5. Nature 1987, 328, 614–616. [Google Scholar] [CrossRef]
- He, T.-C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C- MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive Transcriptional Activation by a β-Catenin-Tcf Complex in APC −/− Colon Carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [Green Version]
- Friedl, W.; Aretz, S. Familial Adenomatous Polyposis: Experience from a Study of 1164 Unrelated German Polyposis Patients. Hered. Cancer Clin. Pract. 2005, 3, 95. [Google Scholar] [CrossRef] [Green Version]
- Nagase, H.; Nakamura, Y. Mutations of TheAPC Adenomatous Polyposis Coli) Gene. Hum. Mutat. 1993, 2, 425–434. [Google Scholar] [CrossRef]
- Plawski, A. Novel Germline Mutations in the Adenomatous Polyposis Coli Gene in Polish Families with Familial Adenomatous Polyposis. J. Med. Genet. 2004, 41, e11. [Google Scholar] [CrossRef] [Green Version]
- Truta, B.; Allen, B.A.; Conrad, P.G.; Weinberg, V.; Miller, G.A.; Pomponio, R.; Lipton, L.R.; Guerra, G.; Tomlinson, I.P.M.; Sleisenger, M.H.; et al. A Comparison of the Phenotype and Genotype in Adenomatous Polyposis Patients with and without a Family History. Fam. Cancer 2005, 4, 127–133. [Google Scholar] [CrossRef]
- Aoun, R.J.N.; Kalady, M.F. The Importance of Genetics for Timing and Extent of Surgery in Inherited Colorectal Cancer Syndromes. Surg. Oncol. 2022, 43, 101765. [Google Scholar] [CrossRef]
- Nagase, H.; Miyoshi, Y.; Horii, A.; Aoki, T.; Ogawa, M.; Utsunomiya, J.; Baba, S.; Sasazuki, T.; Nakamura, Y. Correlation between the Location of Germ-Line Mutations in the APC Gene and the Number of Colorectal Polyps in Familial Adenomatous Polyposis Patients. Cancer Res. 1992, 52, 4055–4057. [Google Scholar]
- Goss, K.H.; Groden, J. Biology of the Adenomatous Polyposis Coli Tumor Suppressor. J. Clin. Oncol. 2000, 18, 1967–1979. [Google Scholar] [CrossRef]
- Friedl, W. Can APC Mutation Analysis Contribute to Therapeutic Decisions in Familial Adenomatous Polyposis? Experience from 680 FAP Families. Gut 2001, 48, 515–521. [Google Scholar] [CrossRef] [Green Version]
- Ficari, F.; Cama, A.; Valanzano, R.; Curia, M.C.; Palmirotta, R.; Aceto, G.; Esposito, D.L.; Crognale, S.; Lombardi, A.; Messerini, L.; et al. APC Gene Mutations and Colorectal Adenomatosis in Familial Adenomatous Polyposis. Br. J. Cancer 2000, 82, 348–353. [Google Scholar] [CrossRef]
- Walon, C.; Kartheuser, A.; Michils, G.; Smaers, M.; Lannoy, N.; Ngounou, P.; Mertens, G.; Verellen-Dumoulin, C. Novel Germline Mutations in the APC Gene and Their Phenotypic Spectrum in Familial Adenomatous Polyposis Kindreds. Hum. Genet. 1997, 100, 601–605. [Google Scholar] [CrossRef]
- Gebert, J.F.; Dupon, C.; Kadmon, M.; Hahn, M.; Herfarth, C.; von Knebel Doeberitz, M.; Schackert, H.K. Combined Molecular and Clinical Approaches for the Identification of Families with Familial Adenomatous Polyposis Coli. Ann. Surg. 1999, 229, 350–361. [Google Scholar] [CrossRef]
- Michils, G.; Tejpar, S.; Fryns, J.-P.; Legius, E.; Van Cutsem, E.; Cassiman, J.-J.; Matthijs, G. Pathogenic Mutations and Rare Variants of the APC Gene Identified in 75 Belgian Patients with Familial Adenomatous Polyposis by Fluorescent Enzymatic Mutation Detection (EMD). Eur. J. Hum. Genet. 2002, 10, 505–510. [Google Scholar] [CrossRef]
- Burt, R.W.; Leppert, M.F.; Slattery, M.L.; Samowitz, W.S.; Spirio, L.N.; Kerber, R.A.; Kuwada, S.K.; Neklason, D.W.; DiSario, J.A.; Lyon, E.; et al. Genetic Testing and Phenotype in a Large Kindred with Attenuated Familial Adenomatous Polyposis. Gastroenterology 2004, 127, 444–451. [Google Scholar] [CrossRef]
- Soravia, C.; Berk, T.; Madlensky, L.; Mitri, A.; Cheng, H.; Gallinger, S.; Cohen, Z.; Bapat, B. Genotype-Phenotype Correlations in Attenuated Adenomatous Polyposis Coli. Am. J. Hum. Genet. 1998, 62, 1290–1301. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, A.L.; Bisgaard, M.L.; Bülow, S. Attenuated Familial Adenomatous Polyposis (AFAP): A Review of the Literature. Fam. Cancer 2003, 2, 43–55. [Google Scholar] [CrossRef]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R.; et al. Inherited Variants of MYH Associated with Somatic G:C→T:A Mutations in Colorectal Tumors. Nat. Genet. 2002, 30, 227–232. [Google Scholar] [CrossRef]
- Farrington, S.M.; Tenesa, A.; Barnetson, R.; Wiltshire, A.; Prendergast, J.; Porteous, M.; Campbell, H.; Dunlop, M.G. Germline Susceptibility to Colorectal Cancer Due to Base-Excision Repair Gene Defects. Am. J. Hum. Genet. 2005, 77, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Boparai, K.S.; Dekker, E.; van Eeden, S.; Polak, M.M.; Bartelsman, J.F.W.M.; Mathus–Vliegen, E.M.H.; Keller, J.J.; van Noesel, C.J.M. Hyperplastic Polyps and Sessile Serrated Adenomas as a Phenotypic Expression of MYH-Associated Polyposis. Gastroenterology 2008, 135, 2014–2018. [Google Scholar] [CrossRef]
- Wang, L.; Baudhuin, L.M.; Boardman, L.A.; Steenblock, K.J.; Petersen, G.M.; Halling, K.C.; French, A.J.; Johnson, R.A.; Burgart, L.J.; Rabe, K.; et al. MYH Mutations in Patients with Attenuated and Classic Polyposis and with Young-Onset Colorectal Cancer without Polyps. Gastroenterology 2004, 127, 9–16. [Google Scholar] [CrossRef]
- Lubbe, S.J.; Di Bernardo, M.C.; Chandler, I.P.; Houlston, R.S. Clinical Implications of the Colorectal Cancer Risk Associated with MUTYH Mutation. JCO 2009, 27, 3975–3980. [Google Scholar] [CrossRef]
- Beck, S.H.; Jelsig, A.M.; Yassin, H.M.; Lindberg, L.J.; Wadt, K.A.W.; Karstensen, J.G. Intestinal and Extraintestinal Neoplasms in Patients with NTHL1 Tumor Syndrome: A Systematic Review. Fam. Cancer 2022, 21, 453–462. [Google Scholar] [CrossRef]
- Weren, R.D.A.; Ligtenberg, M.J.L.; Kets, C.M.; de Voer, R.M.; Verwiel, E.T.P.; Spruijt, L.; van Zelst-Stams, W.A.G.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A Germline Homozygous Mutation in the Base-Excision Repair Gene NTHL1 Causes Adenomatous Polyposis and Colorectal Cancer. Nat. Genet. 2015, 47, 668–671. [Google Scholar] [CrossRef]
- Aihara, H.; Kumar, N.; Thompson, C.C. Diagnosis, Surveillance, and Treatment Strategies for Familial Adenomatous Polyposis: Rationale and Update. Eur. J. Gastroenterol. Hepatol. 2014, 26, 255–262. [Google Scholar] [CrossRef]
- Ng, K.-S.; Gonsalves, S.J.; Sagar, P.M. Ileal-Anal Pouches: A Review of Its History, Indications, and Complications. World J. Gastroenterol. 2019, 25, 4320–4342. [Google Scholar] [CrossRef]
- Connor, T.; McPhillips, M.; Hipwell, M.; Ziolkowski, A.; Oldmeadow, C.; Clapham, M.; Pockney, P.; Lis, E.; Banasiewicz, T.; Pławski, A.; et al. CD36 Polymorphisms and the Age of Disease Onset in Patients with Pathogenic Variants within the Mutation Cluster Region of APC. Hered. Cancer Clin. Pract. 2021, 19, 25. [Google Scholar] [CrossRef]
- Plawski, A.; Banasiewicz, T.; Borun, P.; Kubaszewski, L.; Krokowicz, P.; Skrzypczak-Zielinska, M.; Lubinski, J. Familial Adenomatous Polyposis of the Colon. Hered. Cancer Clin. Pract. 2013, 11, 15. [Google Scholar] [CrossRef] [Green Version]
- Jeghers, H.; McKusick, V.A.; Katz, K.H. Generalized Intestinal Polyposis and Melanin Spots of the Oral Mucosa, Lips and Digits: A Syndrome of Diagnostic Significance. N. Engl. J. Med. 1949, 241, 993–1005. [Google Scholar] [CrossRef] [Green Version]
- Peutz, J.L.A. Over Een Zeer Merkwaardige, Gecombineerde Familiaire Polyposis van de Slijmvliezen van Den Tractus Intestinalis Met Die van de Neuskeelholte En Gepaard Met Eigenaardige Pigmentaties van Huid-En Slijmvliezen. Nederl. Maandschr. Geneesk. 1921, 10, 134–146. [Google Scholar]
- Westerman, A.M.; Entius, M.M.; de Baar, E.; Boor, P.P.; Koole, R.; van Velthuysen, M.L.F.; Offerhaus, G.J.A.; Lindhout, D.; de Rooij, F.W.; Wilson, J.P. Peutz-Jeghers Syndrome: 78-Year Follow-up of the Original Family. Lancet 1999, 353, 1211–1215. [Google Scholar] [CrossRef]
- Jass, J.R. Colorectal Polyposes: From Phenotype to Diagnosis. Pathol. Res. Pract. 2008, 204, 431–447. [Google Scholar] [CrossRef]
- Gammon, A.; Jasperson, K.; Kohlmann, W.; Burt, R.W. Hamartomatous Polyposis Syndromes. Best Pract. Res. Clin. Gastroenterol. 2009, 23, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Hinds, R.; Philp, C.; Hyer, W.; Fell, J.M. Complications of Childhood Peutz-Jeghers Syndrome: Implications for Pediatric Screening. J. Pediatr. Gastroenterol. Nutr. 2004, 39, 219–220. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Brensinger, J.D.; Tersmette, A.C.; Goodman, S.N.; Petersen, G.M.; Booker, S.V.; Cruz–Correa, M.; Offerhaus, J.A. Very High Risk of Cancer in Familial Peutz–Jeghers Syndrome. Gastroenterology 2000, 119, 1447–1453. [Google Scholar] [CrossRef] [Green Version]
- Kopacova, M.; Tacheci, I.; Rejchrt, S.; Bures, J. Peutz-Jeghers Syndrome: Diagnostic and Therapeuticapproach. WJG 2009, 15, 5397. [Google Scholar] [CrossRef]
- Hemminki, A.; Tomlinson, I.; Markie, D.; Järvinen, H.; Sistonen, P.; Björkqvist, A.-M.; Knuutila, S.; Salovaara, R.; Bodmer, W.; Shibata, D.; et al. Localization of a Susceptibility Locus for Peutz-Jeghers Syndrome to 19p Using Comparative Genomic Hybridization and Targeted Linkage Analysis. Nat. Genet. 1997, 15, 87–90. [Google Scholar] [CrossRef]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Höglund, P.; et al. A Serine/Threonine Kinase Gene Defective in Peutz–Jeghers Syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef]
- Jenne, D.E.; Reomann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Müller, O.; Back, W.; Zimmer, M. Peutz-Jeghers Syndrome Is Caused by Mutations in a Novel Serine Threoninekinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef]
- Olschwang, S.; Markie, D.; Seal, S.; Neale, K.; Phillips, R.; Cottrell, S.; Ellis, I.; Hodgson, S.; Zauber, P.; Spigelman, A.; et al. Peutz-Jeghers Disease: Most, but Not All, Families Are Compatible with Linkage to 19p13.3. J. Med. Genet. 1998, 35, 42–44. [Google Scholar] [CrossRef]
- Aretz, S.; Stienen, D.; Uhlhaas, S.; Loff, S.; Back, W.; Pagenstecher, C.; McLeod, D.R.; Graham, G.E.; Mangold, E.; Santer, R.; et al. High Proportion of Large Genomic STK11 Deletions in Peutz-Jeghers Syndrome. Hum. Mutat. 2005, 26, 513–519. [Google Scholar] [CrossRef]
- Borun, P.; Bartkowiak, A.; Banasiewicz, T.; Nedoszytko, B.; Nowakowska, D.; Teisseyre, M.; Limon, J.; Lubinski, J.; Kubaszewski, L.; Walkowiak, J.; et al. High Resolution Melting Analysis as a Rapid and Efficient Method of Screening for Small Mutations in the STK11gene in Patients with Peutz-Jeghers Syndrome. BMC Med. Genet. 2013, 14, 58. [Google Scholar] [CrossRef]
- Borun, P.; De Rosa, M.; Nedoszytko, B.; Walkowiak, J.; Plawski, A. Specific Alu Elements Involved in a Significant Percentage of Copy Number Variations of the STK11 Gene in Patients with Peutz–Jeghers Syndrome. Fam. Cancer 2015, 14, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Szmulewicz, M.N.; Novick, G.E.; Herrera, R.J. Effects OfAlu Insertions on Gene Function. Electrophoresis 1998, 19, 1260–1264. [Google Scholar] [CrossRef]
- de Leng, W.; Jansen, M.; Carvalho, R.; Polak, M.; Musler, A.; Milne, A.; Keller, J.; Menko, F.; de Rooij, F.; Iacobuzio-Donahue, C.; et al. Genetic Defects Underlying Peutz-Jeghers Syndrome (PJS) and Exclusion of the Polarity-Associated MARK/Par1 Gene Family as Potential PJS Candidates. Clin. Genet. 2007, 72, 568–573. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.-L.; Zhao, Z.-Y.; Li, B.-R.; Yang, F.; Li, J.; Jin, X.-W.; Wang, H.; Yu, E.-D.; Sun, S.-H.; Ning, S.-B. The Altered Activity of P53 Signaling Pathway by STK11 Gene Mutations and Its Cancer Phenotype in Peutz-Jeghers Syndrome. BMC Med. Genet. 2018, 19, 141. [Google Scholar] [CrossRef] [Green Version]
- Karuman, P.; Gozani, O.; Odze, R.D.; Zhou, X.C.; Zhu, H.; Shaw, R.; Brien, T.P.; Bozzuto, C.D.; Ooi, D.; Cantley, L.C.; et al. The Peutz-Jegher Gene Product LKB1 Is a Mediator of P53-Dependent Cell Death. Mol. Cell 2001, 7, 1307–1319. [Google Scholar] [CrossRef]
- Ingrosso, M.; Prete, F.; Pisani, A.; Carbonara, R.; Azzarone, A.; Francavilla, A. Laparoscopically Assisted Total Enteroscopy: A New Approach to Small Intestinal Diseases. Gastrointest. Endosc. 1999, 49, 651–653. [Google Scholar] [CrossRef]
- Brüggmann, D.; Tchartchian, G.; Wallwiener, M.; Münstedt, K.; Tinneberg, H.-R.; Hackethal, A. Intra-Abdominal Adhesions. Dtsch. Ärzteblatt Int. 2010, 107, 769–775. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Sekine, Y.; Sato, Y.; Higashizawa, T.; Miyata, T.; Iino, S.; Ido, K.; Sugano, K. Total Enteroscopy with a Nonsurgical Steerable Double-Balloon Method. Gastrointest. Endosc. 2001, 53, 216–220. [Google Scholar] [CrossRef]
- Robinson, J.; Lai, C.; Martin, A.; Nye, E.; Tomlinson, I.; Silver, A. Oral Rapamycin Reduces Tumour Burden and Vascularization in Lkb1 +/− Mice: Rapamycin in Lkb1 +/− Mice. J. Pathol. 2009, 219, 35–40. [Google Scholar] [CrossRef]
- Wei, C.; Amos, C.I.; Zhang, N.; Zhu, J.; Wang, X.; Frazier, M.L. Chemopreventive Efficacy of Rapamycin on Peutz–Jeghers Syndrome in a Mouse Model. Cancer Lett. 2009, 277, 149–154. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Hamilton, S.R.; Kern, S.E.; Offerhaus, G.J.; Green, P.A.; Celano, P.; Krush, A.J.; Booker, S.V. Colorectal Neoplasia in Juvenile Polyposis or Juvenile Polyps. Arch. Dis. Child. 1991, 66, 971–975. [Google Scholar] [CrossRef] [Green Version]
- Howe, J.R.; Mitros, F.A.; Summers, R.W. The Risk of Gastrointestinal Carcinoma in Familial Juvenile Polyposis. Ann. Surg. Oncol. 1998, 5, 751–756. [Google Scholar] [CrossRef]
- Brosens, L.A.A.; van Hattem, A.; Hylind, L.M.; Iacobuzio-Donahue, C.; Romans, K.E.; Axilbund, J.; Cruz-Correa, M.; Tersmette, A.C.; Offerhaus, G.J.A.; Giardiello, F.M. Risk of Colorectal Cancer in Juvenile Polyposis. Gut 2007, 56, 965–967. [Google Scholar] [CrossRef] [Green Version]
- Jass, J.R.; Williams, C.B.; Bussey, H.J.; Morson, B.C. Juvenile Polyposis--a Precancerous Condition. Histopathology 1988, 13, 619–630. [Google Scholar] [CrossRef]
- Blatter, R.; Tschupp, B.; Aretz, S.; Bernstein, I.; Colas, C.; Evans, D.G.; Genuardi, M.; Hes, F.J.; Hüneburg, R.; Järvinen, H.; et al. Disease Expression in Juvenile Polyposis Syndrome: A Retrospective Survey on a Cohort of 221 European Patients and Comparison with a Literature-Derived Cohort of 473 SMAD4/BMPR1A Pathogenic Variant Carriers. Genet. Med. 2020, 22, 1524–1532. [Google Scholar] [CrossRef]
- Hamilton, S.R.; Aaltonen, L.A. Pathology and Genetics of Tumours of the Digestive System; IARC Press: Lyon, France, 2000; ISBN 978-92-832-2410-5. [Google Scholar]
- Aretz, S.; Stienen, D.; Uhlhaas, S.; Stolte, M.; Entius, M.M.; Loff, S.; Back, W.; Kaufmann, A.; Keller, K.-M.; Blaas, S.H.; et al. High Proportion of Large Genomic Deletions and a Genotype Phenotype Update in 80 Unrelated Families with Juvenile Polyposis Syndrome. J. Med. Genet. 2007, 44, 702–709. [Google Scholar] [CrossRef] [Green Version]
- He, X.C.; Zhang, J.; Tong, W.-G.; Tawfik, O.; Ross, J.; Scoville, D.H.; Tian, Q.; Zeng, X.; He, X.; Wiedemann, L.M.; et al. BMP Signaling Inhibits Intestinal Stem Cell Self-Renewal through Suppression of Wnt–β-Catenin Signaling. Nat. Genet. 2004, 36, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Buckhaults, P.; Zawel, L.; Bunz, F.; Riggins, G.; Le Dai, J.; Kern, S.E.; Kinzler, K.W.; Vogelstein, B. Targeted Deletion of Smad4 Shows It Is Required for Transforming Growth Factor β and Activin Signaling in Colorectal Cancer Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 2412–2416. [Google Scholar] [CrossRef] [Green Version]
- van Hattem, W.A.; Langeveld, D.; de Leng, W.W.J.; Morsink, F.H.; van Diest, P.J.; Iacobuzio-Donahue, C.A.; Giardiello, F.M.; Offerhaus, G.J.A.; Brosens, L.A.A. Histologic Variations in Juvenile Polyp Phenotype Correlate with Genetic Defect Underlying Juvenile Polyposis. Am. J. Surg. Pathol. 2011, 35, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Friedl, W.; Uhlhaas, S.; Schulmann, K.; Stolte, M.; Loff, S.; Back, W.; Mangold, E.; Stern, M.; Knaebel, H.-P.; Sutter, C.; et al. Juvenile Polyposis: Massive Gastric Polyposis Is More Common in MADH4 Mutation Carriers than in BMPR1A Mutation Carriers. Hum. Genet. 2002, 111, 108–111. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Landscaping the Cancer Terrain. Science 1998, 280, 1036–1037. [Google Scholar] [CrossRef]
- Haramis, A.-P.G.; Begthel, H.; van den Born, M.; van Es, J.; Jonkheer, S.; Offerhaus, G.J.A.; Clevers, H. De Novo Crypt Formation and Juvenile Polyposis on BMP Inhibition in Mouse Intestine. Science 2004, 303, 1684–1686. [Google Scholar] [CrossRef] [Green Version]
- Woodford-Richens, K.; Williamson, J.; Bevan, S.; Young, J.; Leggett, B.; Frayling, I.; Thway, Y.; Hodgson, S.; Kim, J.C.; Iwama, T.; et al. Allelic Loss at SMAD4 in Polyps from Juvenile Polyposis Patients and Use of Fluorescence in Situ Hybridization to Demonstrate Clonal Origin of the Epithelium. Cancer Res. 2000, 60, 2477–2482. [Google Scholar]
- Oncel, M.; Church, J.M.; Remzi, F.H.; Fazio, V.W. Colonic Surgery in Patients with Juvenile Polyposis Syndrome: A Case Series. Dis. Colon Rectum 2005, 48, 49–56. [Google Scholar] [CrossRef]
- Scott-Conner, C.E.; Hausmann, M.; Hall, T.J.; Skelton, D.S.; Anglin, B.L.; Subramony, C. Familial Juvenile Polyposis: Patterns of Recurrence and Implications for Surgical Management. J. Am. Coll. Surg. 1995, 181, 407–413. [Google Scholar]
- van Hattem, W.A.; Brosens, L.A.A.; Marks, S.Y.; Milne, A.N.A.; van Eeden, S.; Iacobuzio–Donahue, C.A.; Ristimäki, A.; Giardiello, F.M.; Offerhaus, G.J.A. Increased Cyclooxygenase-2 Expression in Juvenile Polyposis Syndrome. Clin. Gastroenterol. Hepatol. 2009, 7, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, K.M. Cowden’s Disease: A Possible New Symptom Complex with Multiple System Involvement. Ann. Intern. Med. 1963, 58, 136. [Google Scholar] [CrossRef]
- Starink, T.M.; van der Veen, J.P.; Arwert, F.; de Waal, L.P.; de Lange, G.G.; Gille, J.J.; Eriksson, A.W. The Cowden Syndrome: A Clinical and Genetic Study in 21 Patients. Clin. Genet. 1986, 29, 222–233. [Google Scholar] [CrossRef]
- Hansen-Kiss, E.; Beinkampen, S.; Adler, B.; Frazier, T.; Prior, T.; Erdman, S.; Eng, C.; Herman, G. A Retrospective Chart Review of the Features of PTEN Hamartoma Tumour Syndrome in Children. J. Med. Genet. 2017, 54, 471–478. [Google Scholar] [CrossRef]
- Tan, M.-H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime Cancer Risks in Individuals with Germline PTEN Mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef] [Green Version]
- Nelen, M.R.; Kremer, H.; Konings, I.B.; Schoute, F.; van Essen, A.J.; Koch, R.; Woods, C.G.; Fryns, J.-P.; Hamel, B.; Hoefsloot, L.H.; et al. Novel PTEN Mutations in Patients with Cowden Disease: Absence of Clear Genotype–Phenotype Correlations. Eur J. Hum. Genet. 1999, 7, 267–273. [Google Scholar] [CrossRef]
- Yehia, L.; Eng, C. PTEN Hamartoma Tumor Syndrome. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Nelen, M.R.; Padberg, G.W.; Peeters, E.A.J.; Lin, A.Y.; van den Helm, B.; Frants, R.R.; Goulon, V.; Goldstein, A.M.; van Reen, M.M.M.; Eastern, D.F.; et al. Localization of the Gene for Cowden Disease to Chromosome 10q22–23. Nat. Genet. 1996, 13, 114–116. [Google Scholar] [CrossRef]
- Dahia, P. PTEN Is Inversely Correlated with the Cell Survival Factor Akt/PKB and Is Inactivated via Multiple Mechanismsin Haematological Malignancies. Hum. Mol. Genet. 1999, 8, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Gorlin, R.J.; Cohen, M.M.; Condon, L.M.; Burke, B.A. Bannayan-Riley-Ruvalcaba Syndrome. Am. J. Med. Genet. 1992, 44, 307–314. [Google Scholar] [CrossRef]
- Zhou, X.-P.; Hampel, H.; Thiele, H.; Gorlin, R.J.; Hennekam, R.C.; Parisi, M.; Winter, R.M.; Eng, C. Association of Germline Mutation in the PTEN Tumour Suppressor Gene and Proteus and Proteus-like Syndromes. Lancet 2001, 358, 210–211. [Google Scholar] [CrossRef]
- Tan, M.-H.; Mester, J.; Peterson, C.; Yang, Y.; Chen, J.-L.; Rybicki, L.A.; Milas, K.; Pederson, H.; Remzi, B.; Orloff, M.S.; et al. A Clinical Scoring System for Selection of Patients for PTEN Mutation Testing Is Proposed on the Basis of a Prospective Study of 3042 Probands. Am. J. Hum. Genet. 2011, 88, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Crivelli, L.; Bubien, V.; Jones, N.; Chiron, J.; Bonnet, F.; Barouk-Simonet, E.; Couzigou, P.; Sevenet, N.; Caux, F.; Longy, M. Insertion of Alu Elements at a PTEN Hotspot in Cowden Syndrome. Eur. J. Hum. Genet. 2017, 25, 1087–1091. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.-P.; Waite, K.A.; Pilarski, R.; Hampel, H.; Fernandez, M.J.; Bos, C.; Dasouki, M.; Feldman, G.L.; Greenberg, L.A.; Ivanovich, J.; et al. Germline PTEN Promoter Mutations and Deletions in Cowden/Bannayan-Riley-Ruvalcaba Syndrome Result in Aberrant PTEN Protein and Dysregulation of the Phosphoinositol-3-Kinase/Akt Pathway. Am. J. Hum. Genet. 2003, 73, 404–411. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.J.; Romigh, T.; Sesock, K.; Eng, C. Characterization of Cryptic Splicing in Germline PTEN Intronic Variants in Cowden Syndrome. Hum. Mutat. 2017, 38, 1372–1377. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.; Zbuk, K.M.; Sadler, T.; Patocs, A.; Lobo, G.; Edelman, E.; Platzer, P.; Orloff, M.S.; Waite, K.A.; Eng, C. Germline Mutations and Variants in the Succinate Dehydrogenase Genes in Cowden and Cowden-like Syndromes. Am. J. Hum. Genet. 2008, 83, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, L.C.; Schaid, D.J.; Woods, J.E.; Crotty, T.P.; Myers, J.L.; Arnold, P.G.; Petty, P.M.; Sellers, T.A.; Johnson, J.L.; McDonnell, S.K.; et al. Efficacy of Bilateral Prophylactic Mastectomy in Women with a Family History of Breast Cancer. N. Engl. J. Med. 1999, 340, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Squarize, C.H.; Castilho, R.M.; Gutkind, J.S. Chemoprevention and Treatment of Experimental Cowden’s Disease by MTOR Inhibition with Rapamycin. Cancer Res. 2008, 68, 7066–7072. [Google Scholar] [CrossRef]
- Biller, L.H.; Syngal, S.; Yurgelun, M.B. Recent Advances in Lynch Syndrome. Fam. Cancer 2019, 18, 211–219. [Google Scholar] [CrossRef]
- Croner, R.S.; Brueckl, W.M.; Reingruber, B.; Hohenberger, W.; Guenther, K. Age and Manifestation Related Symptoms in Familial Adenomatous Polyposis. BMC Cancer 2005, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Eccles, D.M.; Lunt, P.W.; Wallis, Y.; Griffiths, M.; Sandhu, B.; McKay, S.; Morton, D.; Shea-Simonds, J.; MacDonald, F. An Unusually Severe Phenotype for Familial Adenomatous Polyposis. Arch. Dis. Child. 1997, 77, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.; Barnes, D.R.; Dunlop, J.; Barrowdale, D.; Antoniou, A.C.; Berg, J.N. Attenuated Familial Adenomatous Polyposis Manifests as Autosomal Dominant Late-Onset Colorectal Cancer. Eur. J. Hum. Genet. 2014, 22, 1330–1333. [Google Scholar] [CrossRef]
- Kuiper, R.P.; Nielsen, M.; De Voer, R.M.; Hoogerbrugge, N. NTHL1 Tumor Syndrome. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Menahem, B.; Alves, A.; Regimbeau, J.M.; Sabbagh, C. Lynch Syndrome: Current Management In 2019. J. Visc. Surg. 2019, 156, 507–514. [Google Scholar] [CrossRef]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. ACG Clinical Guideline: Genetic Testing and Management of Hereditary Gastrointestinal Cancer Syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef] [Green Version]
- Galiatsatos, P.; Foulkes, W.D. Familial Adenomatous Polyposis. Am. J. Gastroenterol. 2006, 101, 385–398. [Google Scholar] [CrossRef]
- Vogt, S.; Jones, N.; Christian, D.; Engel, C.; Nielsen, M.; Kaufmann, A.; Steinke, V.; Vasen, H.F.; Propping, P.; Sampson, J.R.; et al. Expanded Extracolonic Tumor Spectrum in MUTYH-Associated Polyposis. Gastroenterology 2009, 137, 1976–1985.e10. [Google Scholar] [CrossRef]
- Win, A.K.; Reece, J.C.; Dowty, J.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Southey, M.C.; Young, J.P.; Cleary, S.P.; Kim, H.; et al. Risk of Extracolonic Cancers for People with Biallelic and Monoallelic Mutations in MUTYH: Extracolonic Cancer Risks for People with Biallelic and Monoallelic MUTYH Mutations. Int. J. Cancer 2016, 139, 1557–1563. [Google Scholar] [CrossRef] [Green Version]
- Yen, T.; Stanich, P.P.; Axell, L.; Patel, S.G. APC-Associated Polyposis Conditions. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, Seattle, WA, USA, 1993. [Google Scholar]
- Stanich, P.P. Colonic Manifestations of PTEN Hamartoma Tumor Syndrome: Case Series and Systematic Review. WJG 2014, 20, 1833. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Syndrome | Gene Mutations |
|---|---|
| LS | MSH2, MLH1, MSH6, PMS2 |
| CMMRDS | MLH1, MSH2, MSH6, PMS2 |
| FAP | APC |
| AFAP | APC |
| MAP | MUTYH (MYH) |
| NAP | NTHL1 |
| PJS | LKB1 (STK11) |
| JPS | SMAD4, BMPR1A |
| CS | PTEN |
| Syndrome | Average Age of Diagnosis | Number of Polyps | Type of Polyps | Location of Tumors |
|---|---|---|---|---|
| LS | About 50 years | From 1 to several | Adenomatous | Endometrium, stomach, bile ducts, urinary tract, ovaries |
| CMMRDS | Before 18 years | From 1 to several | Adenomatous | Lymphatic system, brain and central nervous system, colon, rectum, duodenum, jejunum, ileum, uterus, bladder, ureter |
| FAP | 15 years | Numerous (more than 100, mostly uncountable) | Adenomatous | duodenum, fundus of the stomach, liver, adrenal gland, soft tissues, brain, thyroid, bones |
| AFAP | 20 years | Up to 100 | Adenomatous | - |
| MAP | 30–40 years | Up to 100 | Adenomatous | - |
| NAP | 55 years | Up to 100 | Adenomatous | Breast, reproductive organs, bladder, skin |
| PJS | Several years | Several | Hamartomatous | Pancreas, breast, lungs, ovaries, testicles |
| JPS | 10–30 years | Several | Hamartomatous | - |
| CP | Several years | Few | Hamartomatous | Thyroid, bladder, kidneys, breast, nipples, a body of the uterus |
| Organ/Percentage Risk of Cancers | LS | CMMRDS | FAP | AFAP | MAP | PJS | JPS | CS |
|---|---|---|---|---|---|---|---|---|
| Colon | 50–70% | 25% | Up to 100% | 70% | 43–63% | 39% | 38,7% | 9% |
| Duodenum | — | 8% | 3–5% | 4–12% | 4% | — | — | — |
| Bladder | — | 1% | — | — | 6–25% | — | — | 3% |
| Stomach | 7% | — | 5% | — | 1% | 29% | — | — |
| Ovary | 9% | — | — | — | 6–14% | 21% | — | — |
| Liver | — | 2% | — | — | — | — | — | |
| Urinary tract | 3% | — | <1–25% | — | — | — | — | — |
| Small intestine | 3% | 8% | 3–10% | 4–12% | — | 13% | — | — |
| Brain | 3% | 53% | 2% | — | — | — | — | — |
| Pancreas | 4% | — | 1.7% | — | — | 36% | — | — |
| Prostate | — | — | — | — | — | — | — | — |
| Breast | — | — | — | — | — | 54% | — | 82.5% |
| Thyroid | — | — | 2% | 1–2% | — | — | — | 35.2% |
| Uterus | 40–60% | 4% | — | — | — | 9% | — | 28.2% |
| Cervix | — | — | — | — | — | 10% | — | — |
| Testis | — | — | — | — | — | 9% | — | — |
| Lungs | — | — | — | — | — | 15% | — | — |
| Skin | — | — | — | — | — | — | — | 6% |
| Lymphatic system | — | 31% | — | — | — | — | — | — |
| Organ/Average Age of Diagnosis of Cancers | LS | CMMRDS | FAP | AFAP | MAP | PJS | JPS | CS |
|---|---|---|---|---|---|---|---|---|
| Colon | 45 | 16 | 40 | 55 | 40–60 | 45.8 | 43.9 | 47 |
| Duodenum | — | 28 | 44 | 60 | 61 | — | — | — |
| Bladder | — | 20 | — | — | 61 | — | — | — |
| Stomach | 49–55 | — | 49 | — | 38 | 30.1 | 54 | — |
| Ovary | 42–54 | — | — | — | 51 | 28 | — | — |
| Liver | 54–57 | — | <5 | — | — | — | — | — |
| Urinary tract | 52–57 | — | — | — | — | — | 40 | |
| Small intestine | 46–51 | 28 | 44 | 60 | — | 41.7 | — | — |
| Brain | 50–55 | 9 | 15–21 | — | — | — | — | — |
| Pancreas | 51.5–56.5 | — | 50 | — | — | 40.8 | — | — |
| Prostate | 59–60 | — | — | — | — | — | — | — |
| Breast | 46–52 | — | — | — | — | 37 | — | 38–46 |
| Thyroid | — | — | 25–33 | 26 | — | — | — | 31–38 |
| Uterus | — | 28 | — | — | — | 43 | — | — |
| Cervix | — | — | — | — | — | 34.3 | — | — |
| Testis | — | — | — | — | — | 8.6 | — | — |
| Lungs | — | — | — | — | — | 47 | — | — |
| Lymphatic system | — | 6 | — | — | — | — | — | — |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hryhorowicz, S.; Kaczmarek-Ryś, M.; Lis-Tanaś, E.; Porowski, J.; Szuman, M.; Grot, N.; Kryszczyńska, A.; Paszkowski, J.; Banasiewicz, T.; Pławski, A. Strong Hereditary Predispositions to Colorectal Cancer. Genes 2022, 13, 2326. https://doi.org/10.3390/genes13122326
Hryhorowicz S, Kaczmarek-Ryś M, Lis-Tanaś E, Porowski J, Szuman M, Grot N, Kryszczyńska A, Paszkowski J, Banasiewicz T, Pławski A. Strong Hereditary Predispositions to Colorectal Cancer. Genes. 2022; 13(12):2326. https://doi.org/10.3390/genes13122326
Chicago/Turabian StyleHryhorowicz, Szymon, Marta Kaczmarek-Ryś, Emilia Lis-Tanaś, Jakub Porowski, Marcin Szuman, Natalia Grot, Alicja Kryszczyńska, Jacek Paszkowski, Tomasz Banasiewicz, and Andrzej Pławski. 2022. "Strong Hereditary Predispositions to Colorectal Cancer" Genes 13, no. 12: 2326. https://doi.org/10.3390/genes13122326