
The Expansion of the Spectrum in Stuttering Disorders to a Novel ARMC Gene Family (ARMC3)

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Ethical Approval, Subject Recruitment, and Blood Collection
2.2. Genomic DNA Extraction
2.3. Exome Sequencing
2.4. Variant Search, Classification, and Sanger Sequencing
2.5. Pathogenicity Prediction
2.6. Protein Structure Modeling and Interaction Studies
3. Results
3.1. Clinical Phenotypes of the Patients
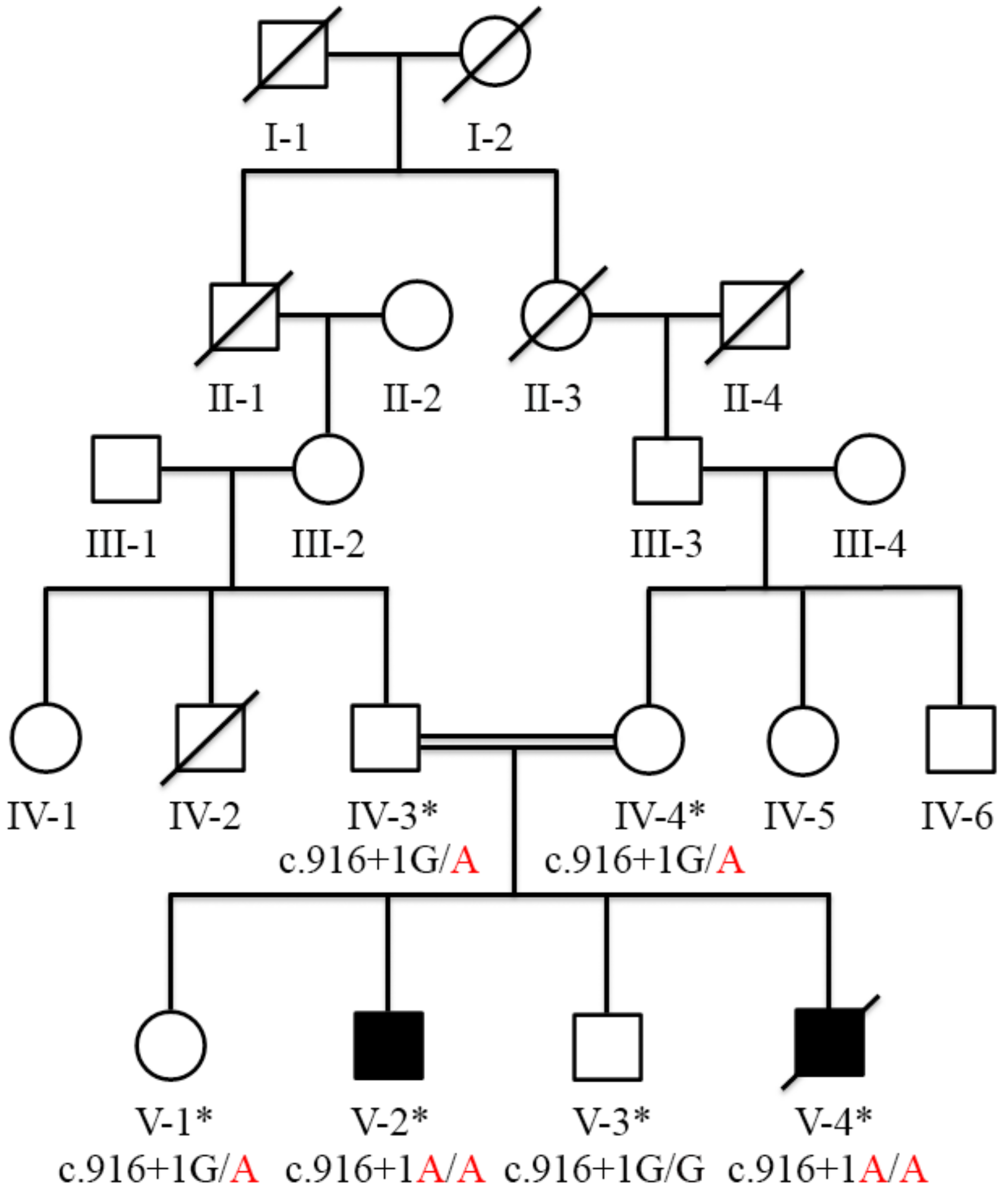
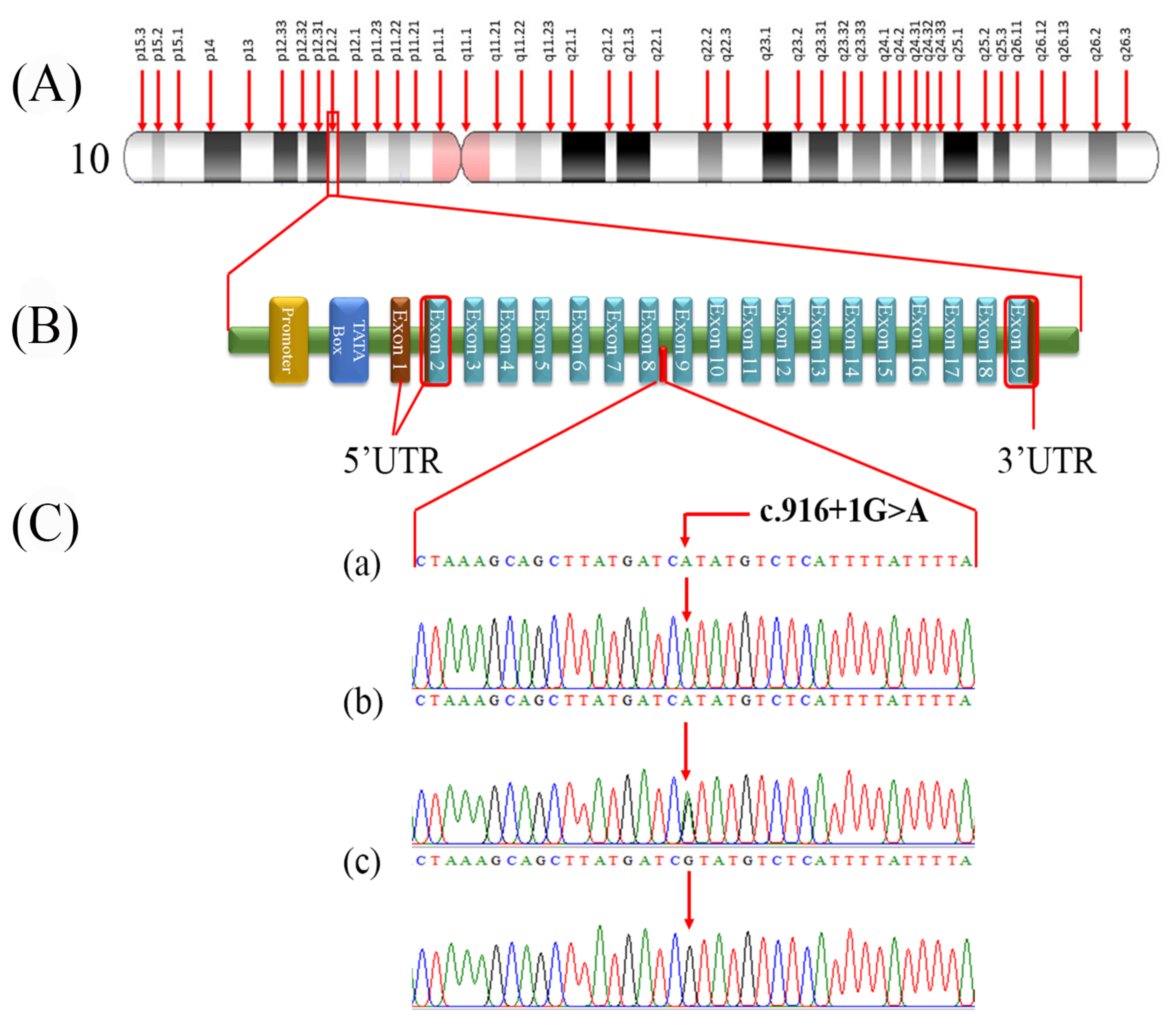
3.2. Variant Identification and Co-Segregation Analysis
3.3. Pathogenicity Prediction
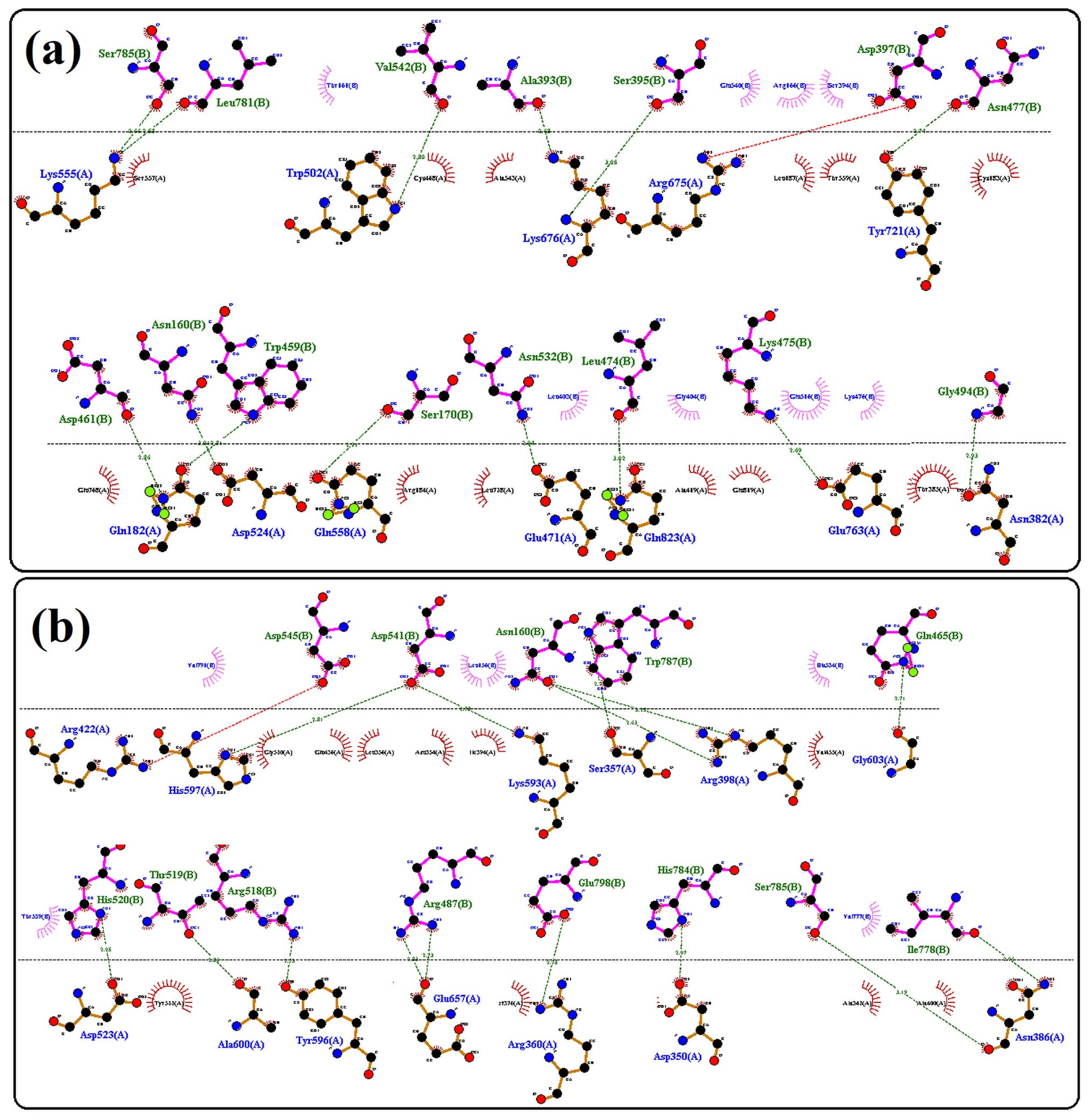
3.4. Protein Structural Findings
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dworzynski, K.; Remington, A.; Rijsdijk, F.; Howell, P.; Plomin, R. Genetic etiology in cases of recovered and persistent stuttering in an unselected, longitudinal sample of young twins. Am. J. Speech Lang. Pathol. 2007, 16, 169–178. [Google Scholar] [CrossRef]
- Yairi, E.; Ambrose, N. Epidemiology of stuttering: 21st century advances. J. Fluen. Disord. 2013, 38, 66–87. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.R.; Haroldson, S.K. Stuttering identification: Standard definition and moment of stuttering. J. Speech. Hear. Res. 1981, 24, 59–63. [Google Scholar]
- Van Borsel, J.; Achten, E.; Santens, P.; Lahorte, P.; Voet, T. fMRI of developmental stuttering: A pilot study. Brain. Lang. 2003, 85, 369–376. [Google Scholar] [CrossRef]
- Månsson, H. Childhood stuttering: Incidence and development. J. Fluen. Disord. 2000, 25, 47–57. [Google Scholar]
- Lundgren, K.; Helm-Estabrooks, N.; Klein, R. Stuttering Following Acquired Brain Damage: A Review of the Literature. J. Neurolinguist. 2010, 23, 447–454. [Google Scholar] [CrossRef] [Green Version]
- Theys, C.; De Nil, L.; Thijs, V.; van Wieringen, A.; Sunaert, S. A crucial role for the cortico-striato-cortical loop in the pathogenesis of stroke-related neurogenic stuttering. Hum. Brain Mapp. 2013, 34, 2103–2112. [Google Scholar] [CrossRef]
- Craig-McQuaide, A.; Akram, H.; Zrinzo, L.; Tripoliti, E. A review of brain circuitries involved in stuttering. Front. Hum. Neurosci. 2014, 8, 884. [Google Scholar] [CrossRef] [Green Version]
- Yairi, E.; Ambrose, N.G.; Paden, E.P.; Throneburg, R.N. Predictive factors of persistence and recovery: Pathways of childhood stuttering. J. Commun. Disord. 1996, 29, 51–77. [Google Scholar] [CrossRef] [Green Version]
- Yairi, E.; Ambrose, N.G. Early childhood stuttering I: Persistency and recovery rates. J. Speech. Lang. Hear. Res. 1999, 42, 1097–1112. [Google Scholar] [CrossRef]
- Frigerio-Domingues, C.; Drayna, D. Genetic contributions to stuttering: The current evidence. Mol. Genet. Genomic. Med. 2017, 5, 95–102. [Google Scholar] [CrossRef]
- Nouri, N.; Nouri, N.; Abdali, H.; Shafie, M.; Karimi, H. Stuttering: Genetic updates and a case report. Adv. Biomed. Res. 2012, 1, 14. [Google Scholar] [CrossRef]
- Sun, Y.; Gao, Y.; Zhou, Y.; Zhou, Y.; Zhang, Y.; Wang, D.; Tan, L.H. IFNAR1 gene mutation may contribute to developmental stuttering in the Chinese population. Hereditas 2021, 158, 46. [Google Scholar] [CrossRef]
- Riaz, N.; Steinberg, S.; Ahmad, J.; Pluzhnikov, A.; Riazuddin, S.; Cox, N.J.; Drayna, D. Genomewide significant linkage to stuttering on chromosome 12. Am. J. Hum. Genet. 2005, 76, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Raza, M.H.; Riazuddin, S.; Drayna, D. Identification of an autosomal recessive stuttering locus on chromosome 3q13.2–3q13.33. Hum. Genet. 2010, 128, 461–463. [Google Scholar] [CrossRef] [Green Version]
- Raza, M.H.; Amjad, R.; Riazuddin, S.; Drayna, D. Studies in a consanguineous family reveal a novel locus for stuttering on chromosome 16q. Hum. Genet. 2012, 131, 311–313. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Riazuddin, S.; Mundorff, J.; Krasnewich, D.; Friedman, P.; Mullikin, J.C.; Drayna, D. Mutations in the lysosomal enzyme-targeting pathway and persistent stuttering. N. Engl. J. Med. 2010, 362, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Raza, M.H.; Mattera, R.; Morell, R.; Sainz, E.; Rahn, R.; Gutierrez, J.; Paris, E.; Root, J.; Solomon, B.; Brewer, C.; et al. Association between Rare Variants in AP4E1, a Component of Intracellular Trafficking, and Persistent Stuttering. Am. J. Hum. Genet. 2015, 97, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Schindler, R.F.; Scotton, C.; Zhang, J.; Passarelli, C.; Ortiz-Bonnin, B.; Simrick, S.; Schwerte, T.; Poon, K.L.; Fang, M.; Rinné, S.; et al. POPDC1(S201F) causes muscular dystrophy and arrhythmia by affecting protein trafficking. J. Clin. Investig. 2016, 126, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Geoffroy, V.; Pizot, C.; Redin, C.; Piton, A.; Vasli, N.; Stoetzel, C.; Blavier, A.; Laporte, J.; Muller, J. VaRank: A simple and powerful tool for ranking genetic variants. PeerJ 2015, 3, e796. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, B.; Ji, C.N.; Kang, Y.; Mao, Y. Cloning and expression of ARMC3_v2, a novel splicing variant of the human ARMC3 gene. Genetika 2006, 42, 999–1003. [Google Scholar]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Coutton, C.; Martinez, G.; Kherraf, Z.E.; Amiri-Yekta, A.; Boguenet, M.; Saut, A.; He, X.; Zhang, F.; Cristou-Kent, M.; Escoffier, J.; et al. Bi-allelic Mutations in ARMC2 Lead to Severe Astheno-Teratozoospermia Due to Sperm Flagellum Malformations in Humans and Mice. Am. J. Hum. Genet. 2019, 104, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Xu, C.; Tan, Q.; Shen, Q.; Wu, H.; Lv, M.; Li, K.; Tang, D.; Song, B.; Xu, Y.; et al. Case Report: Novel Biallelic Mutations in ARMC4 Cause Primary Ciliary Dyskinesia and Male Infertility in a Chinese Family. Front. Genet. 2021, 12, 715339. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Berthon, A. Molecular mechanisms of ARMC5 mutations in adrenal pathophysiology. Curr. Opin. Endocr. Metab. Res. 2019, 8, 104–111. [Google Scholar] [CrossRef]
- Damjanovic, S.S.; Antic, J.A.; Elezovic-Kovacevic, V.I.; Dundjerovic, D.M.; Milicevic, I.T.; Beleslin-Cokic, B.B.; Ilic, B.B.; Rodic, G.S.; Berthon, A.; Maria, A.G.; et al. ARMC5 Alterations in Patients with Sporadic Neuroendocrine Tumors and Multiple Endocrine Neoplasia Type 1 (MEN1). J. Clin. Endocrinol. Metab. 2020, 105, e4531–e4542. [Google Scholar] [CrossRef]
- Kar, A.; Phadke, S.R.; Das Bhowmik, A.; Dalal, A. Whole exome sequencing reveals a mutation in ARMC9 as a cause of mental retardation, ptosis, and polydactyly. Am. J. Med. Genet. A 2018, 176, 34–40. [Google Scholar] [CrossRef]
- Ohno, T.; Meguro, A.; Takeuchi, M.; Yamane, T.; Teshigawara, T.; Kitaichi, N.; Horie, Y.; Namba, K.; Ohno, S.; Nakao, K.; et al. Association Study of ARMC9 Gene Variants with Vogt-Koyanagi-Harada Disease in Japanese Patients. Ocul. Immunol. Inflamm. 2019, 27, 699–705. [Google Scholar] [CrossRef]
- Van De Weghe, J.C.; Rusterholz, T.D.S.; Latour, B.; Grout, M.E.; Aldinger, K.A.; Shaheen, R.; Dempsey, J.C.; Maddirevula, S.; Cheng, Y.H.; Phelps, I.G.; et al. Mutations in ARMC9, which Encodes a Basal Body Protein, Cause Joubert Syndrome in Humans and Ciliopathy Phenotypes in Zebrafish. Am. J. Hum. Genet. 2017, 101, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Park, S.; Miyata, H.; Yu, Z.; Morohoshi, A.; Oura, S.; Matzuk, M.M.; Ikawa, M. ARMC12 regulates spatiotemporal mitochondrial dynamics during spermiogenesis and is required for male fertility. Proc. Natl. Acad. Sci. USA 2021, 118, e2018355118. [Google Scholar] [CrossRef]
- Vlangos, C.N.; Siuniak, A.N.; Robinson, D.; Chinnaiyan, A.M.; Lyons, R.H., Jr.; Cavalcoli, J.D.; Keegan, C.E. Next-generation sequencing identifies the Danforth’s short tail mouse mutation as a retrotransposon insertion affecting Ptf1a expression. PLoS Genet. 2013, 9, e1003205. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Jiang, Z.; Gao, X.; Luo, P.; Jiang, X. ARMC Subfamily: Structures, Functions, Evolutions, Interactions, and Diseases. Front. Mol. Biosci. 2021, 8, 791597. [Google Scholar] [CrossRef]
- Takahashi, H.; Takahashi, K.; Liu, F.C. FOXP genes, neural development, speech and language disorders. Adv. Exp. Med. Biol. 2009, 665, 117–129. [Google Scholar] [CrossRef]
- Richter, G.; Gui, T.; Bourgeois, B.; Koyani, C.N.; Ulz, P.; Heitzer, E.; von Lewinski, D.; Burgering, B.M.T.; Malle, E.; Madl, T. β-catenin regulates FOXP2 transcriptional activity via multiple binding sites. FEBS J. 2021, 288, 3261–3284. [Google Scholar] [CrossRef]
- Vernes, S.C.; Oliver, P.L.; Spiteri, E.; Lockstone, H.E.; Puliyadi, R.; Taylor, J.M.; Ho, J.; Mombereau, C.; Brewer, A.; Lowy, E.; et al. Foxp2 regulates gene networks implicated in neurite outgrowth in the developing brain. PLoS Genet. 2011, 7, e1002145. [Google Scholar] [CrossRef]
- Bonkowsky, J.L.; Wang, X.; Fujimoto, E.; Lee, J.E.; Chien, C.B.; Dorsky, R.I. Domain-specific regulation of foxP2 CNS expression by lef1. BMC Dev. Biol. 2008, 8, 103. [Google Scholar] [CrossRef] [Green Version]
- Abdelmoity, A.T.; Hall, J.J.; Bittel, D.C.; Yu, S. 1.39 Mb inherited interstitial deletion in 12p13.33 associated with developmental delay. Eur. J. Med. Genet. 2011, 54, 198–203. [Google Scholar] [CrossRef]
- Okerlund, N.D.; Cheyette, B.N. Synaptic Wnt signaling-a contributor to major psychiatric disorders? J. Neurodev. Disord. 2011, 3, 162–174. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Participants | Syllables Stuttered | Length of Moment of Stuttering | Avoidance | Speech Tempo and Stutter Frequency | Escape |
|---|---|---|---|---|---|---|
| 1 | IV-3 | NO | NO | NO | Speech tempo: High Stutter frequency: NO | NO |
| 2 | IV-4 | NO | NO | NO | Speech tempo: High Stutter frequency: NO | NO |
| 3 | V-1 | NO | NO | NO | Speech tempo: High Stutter frequency: NO | NO |
| 4 | V-2 | Severely affected | Extremely long | Yes | Speech tempo: Low to Moderate Stutter frequency: High | Continuous extra movements of face or whole body |
| 5 | V-3 | NO | NO | NO | Speech tempo: High Stutter frequency: NO | NO |
| 6 | V-4 | Severely affected | Extremely long | Yes | Speech tempo: Low Stutter frequency: High | Continuous extra movements of face or whole body |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, A.U.; Hamid, M.; Khan, S.A.; Eisa, M.; Ullah, W.; Rehman, Z.U.; Khan, M.A.; Basit, S.; Muhammad, N.; Khan, S.; et al. The Expansion of the Spectrum in Stuttering Disorders to a Novel ARMC Gene Family (ARMC3). Genes 2022, 13, 2299. https://doi.org/10.3390/genes13122299
Rehman AU, Hamid M, Khan SA, Eisa M, Ullah W, Rehman ZU, Khan MA, Basit S, Muhammad N, Khan S, et al. The Expansion of the Spectrum in Stuttering Disorders to a Novel ARMC Gene Family (ARMC3). Genes. 2022; 13(12):2299. https://doi.org/10.3390/genes13122299
Chicago/Turabian StyleRehman, Adil U, Malaika Hamid, Sher Alam Khan, Muhammad Eisa, Wasim Ullah, Zia Ur Rehman, Muzammil Ahmad Khan, Sulman Basit, Noor Muhammad, Saadullah Khan, and et al. 2022. "The Expansion of the Spectrum in Stuttering Disorders to a Novel ARMC Gene Family (ARMC3)" Genes 13, no. 12: 2299. https://doi.org/10.3390/genes13122299