
The Sum of Two Halves May Be Different from the Whole—Effects of Splitting Sequencing Samples Across Lanes

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results
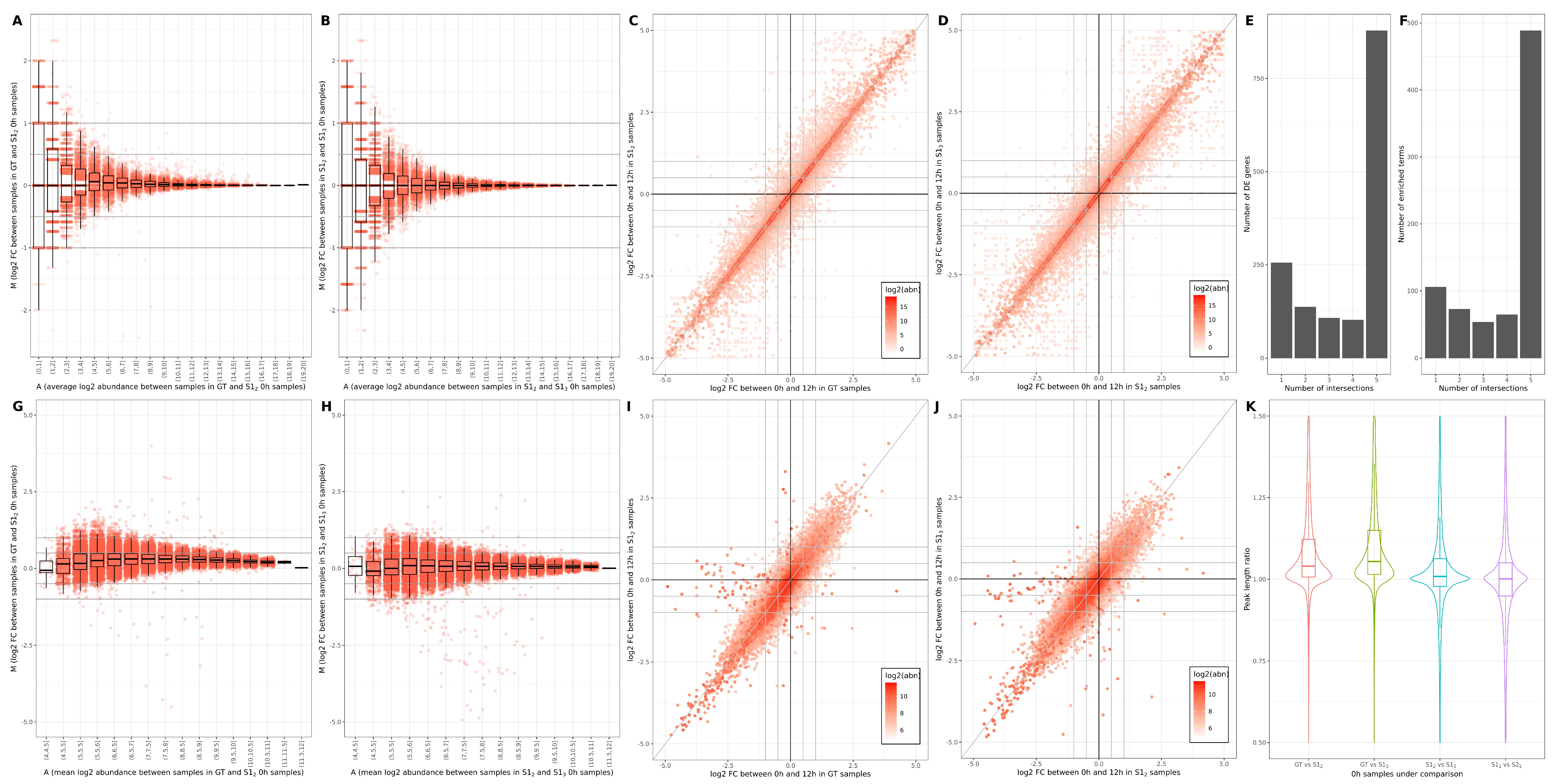
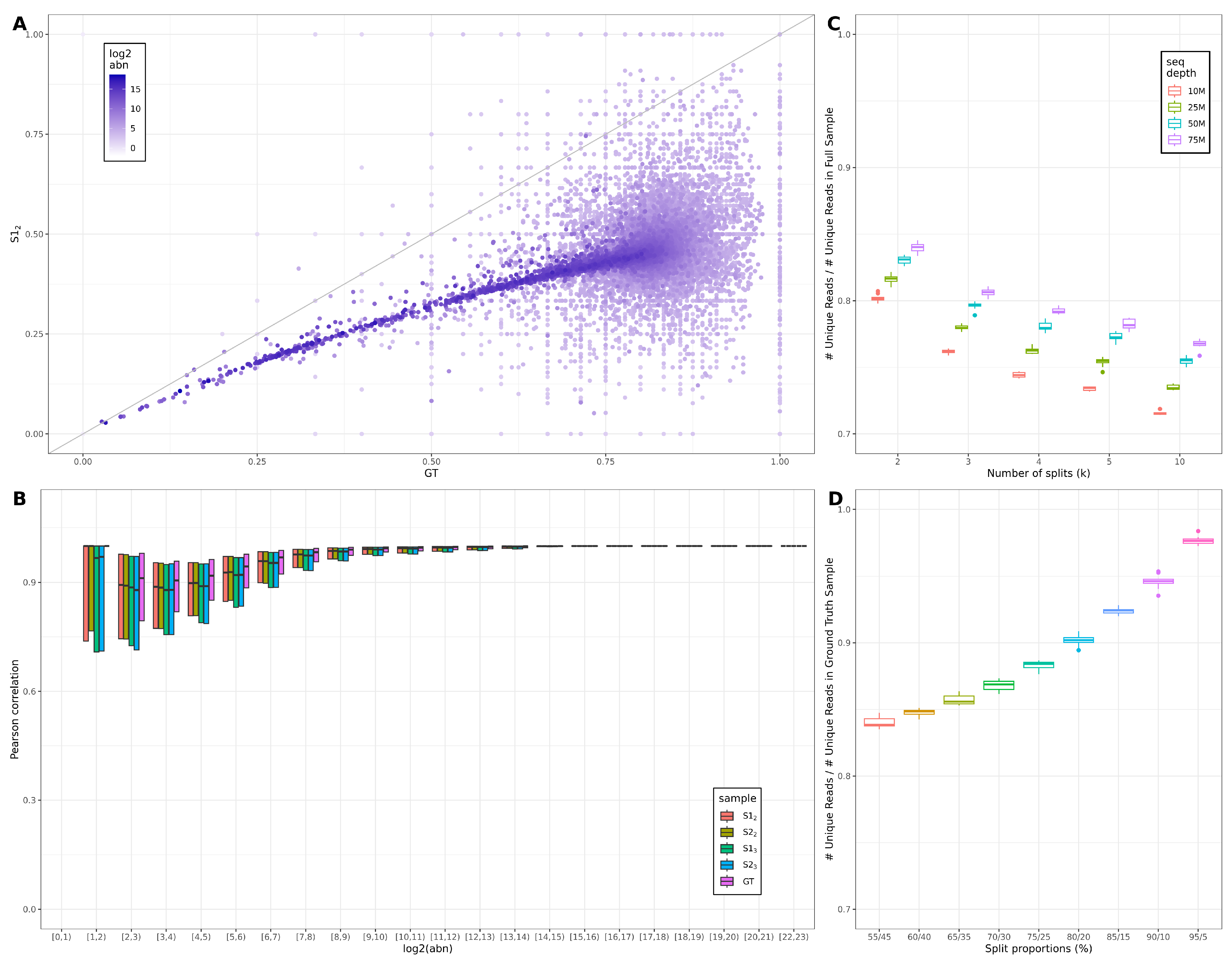
3.1. Across-Lane Split Leads to Differences in Bulk mRNAseq Data
3.2. Consequences of Across-Lane Splitting on Bulk Data
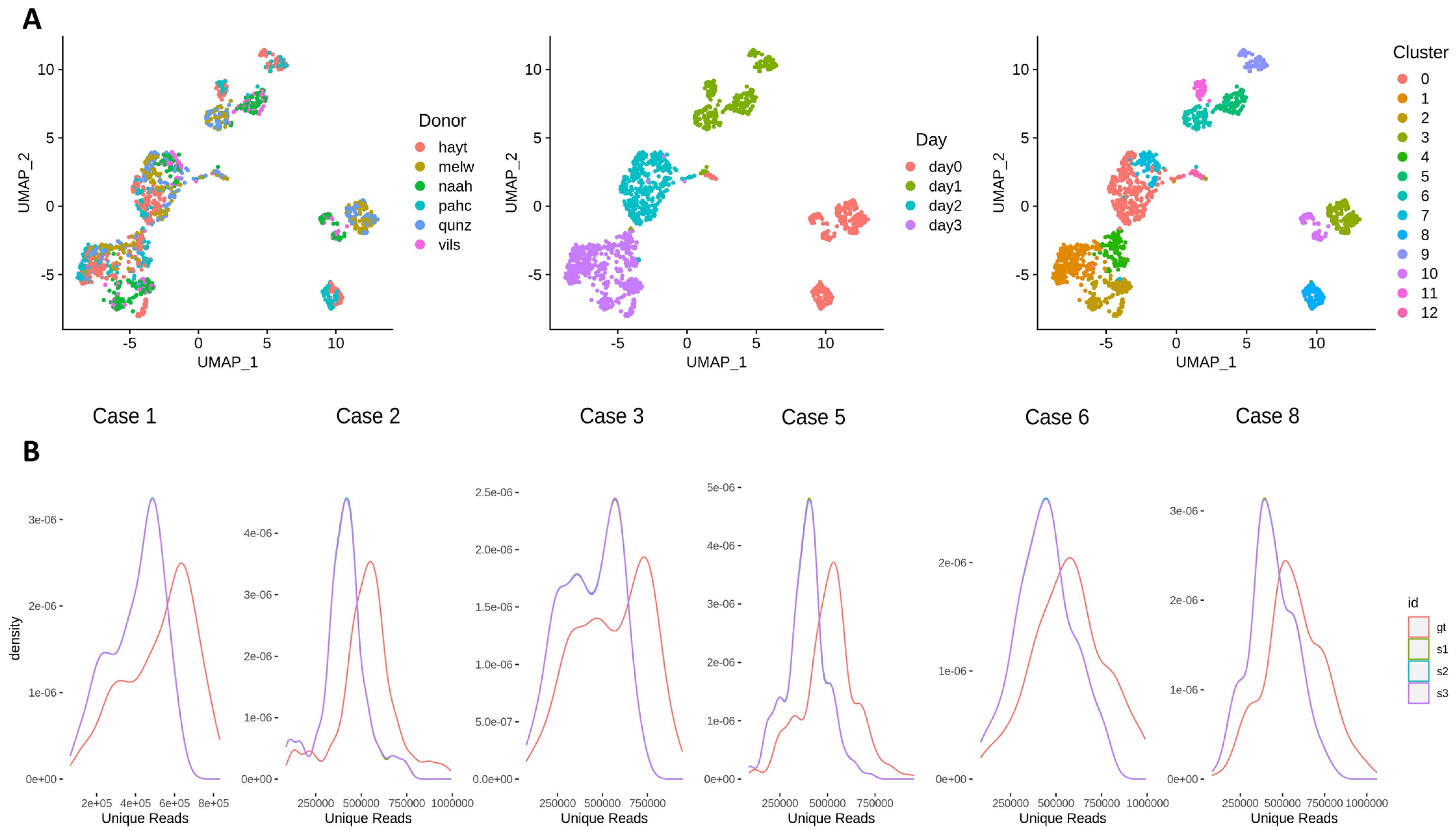
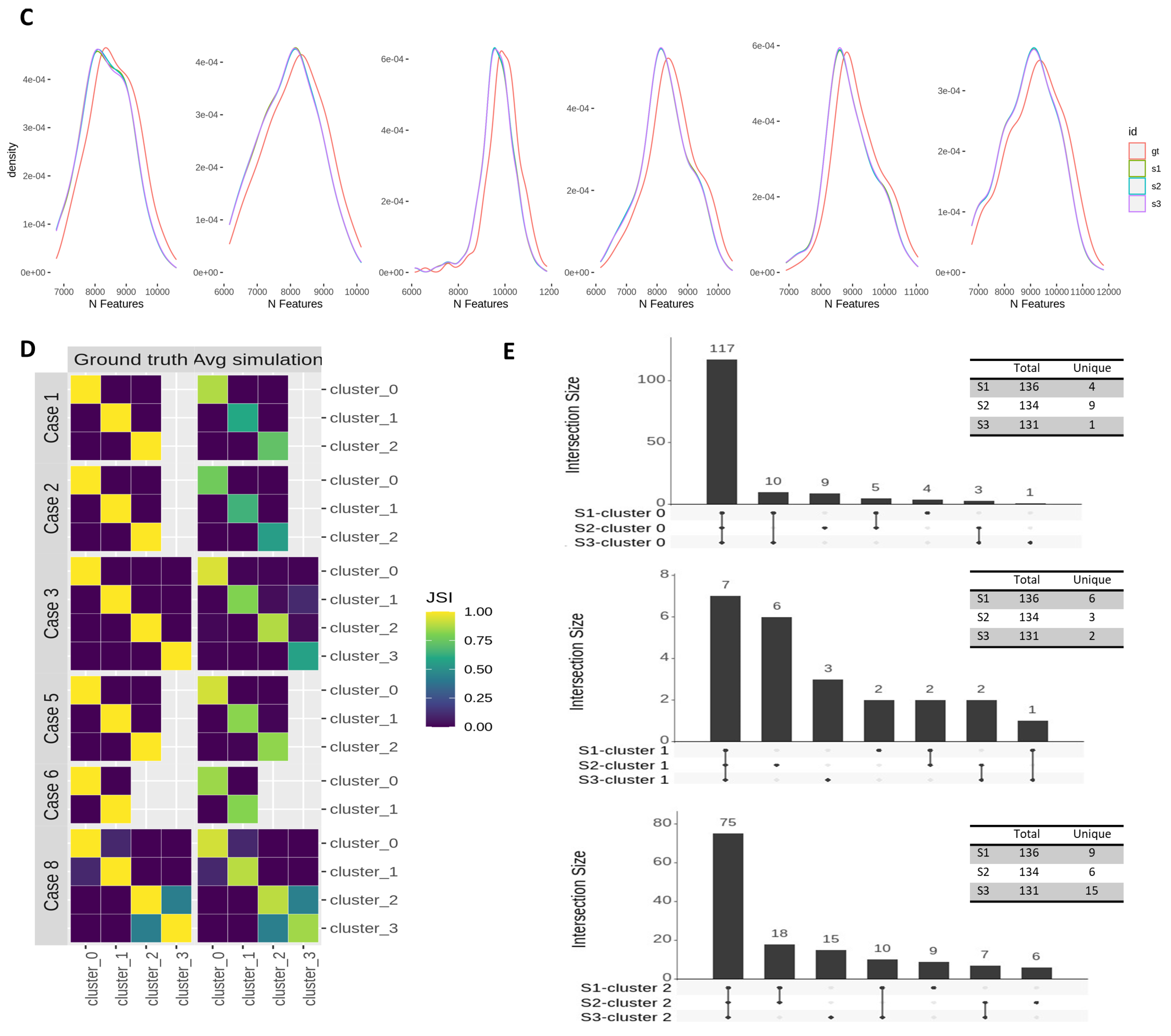
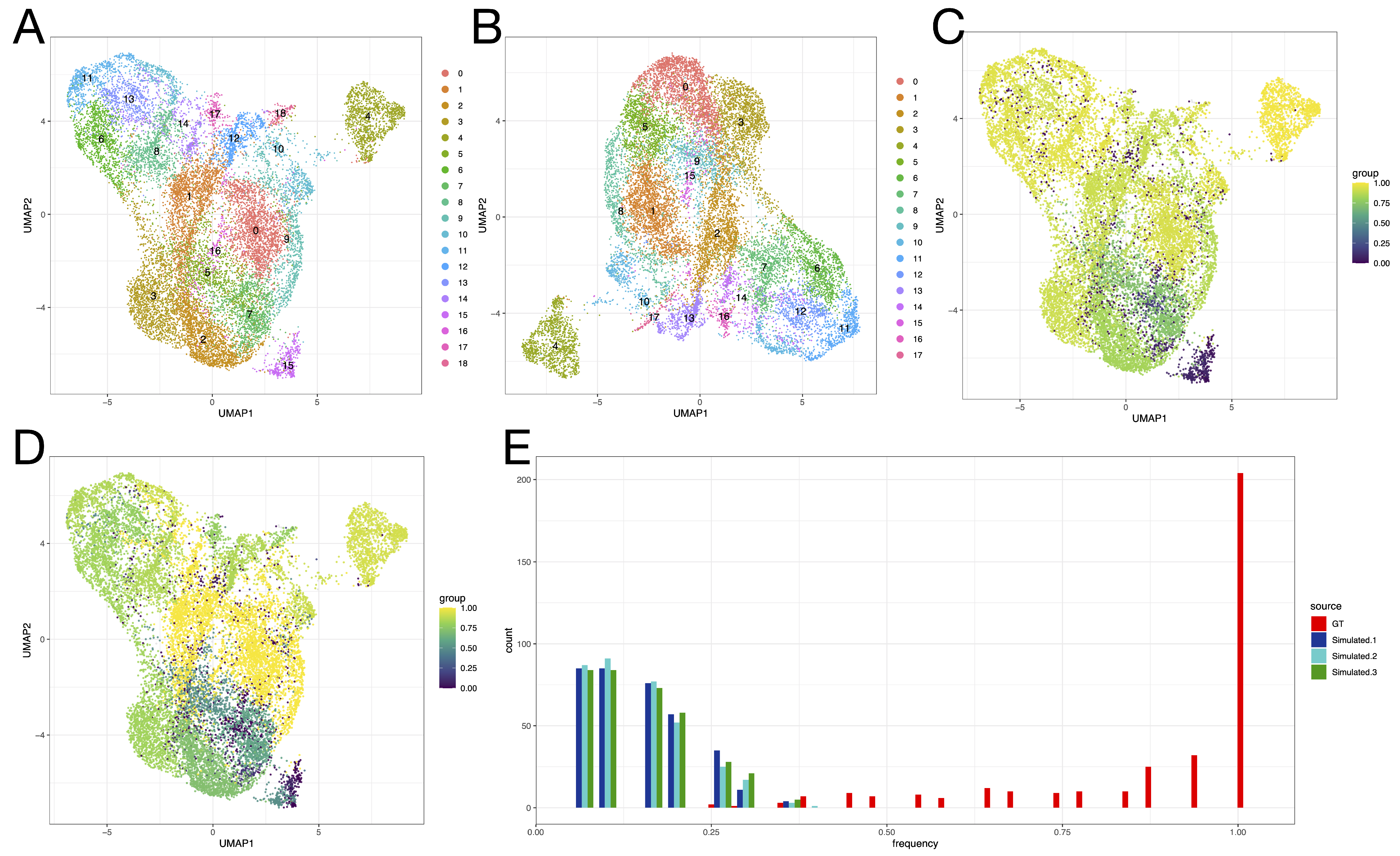
3.3. Effects of Across-Lane Splitting on Single-Cell Data
4. Discussion
4.1. Effects of Splitting on Read Diversity and Levels of Noise
4.2. Effects of Varying the Number and Proportions of Splits
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Yandell, M.; Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012, 13, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Steward, C.; Parker, A.; Minassian, B.; Sisodiya, S.; Frankish, A.; Harrow, J. Genome annotation for clinical genomic diagnostics: Strengths and weaknesses. Genome Med. 2017, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Salzberg, S. Next-generation genome annotation: We still struggle to get it right. Genome Biol. 2019, 20, 92. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Wojciech Szcześniak, M.; Gaffney, D.; Elo, L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [Green Version]
- Oshlack, A.; Robinson, M.; Young, M. From RNA-seq reads to differential expression results. Genome Biol. 2010, 11, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lightbody, G.; Haberland, V.; Browne, F.; Taggart, L.; Zheng, H.; Parkes, E.; Blayney, J. Review of applications of high-throughput sequencing in personalized medicine: Barriers and facilitators of future progress in research and clinical application. Briefings Bioinform. 2019, 20, 1795–1811. [Google Scholar] [CrossRef] [PubMed]
- Lücken, M.; Theis, F. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef]
- McGuire, A.; Gabriel, S.; Tishkoff, S.; Wonkam, A.; Chakravarti, A.; Furlong, E.; Treutlein, B.; Meissner, A.; Chang, H.; López-Bigas, N.; et al. The road ahead in genetics and genomics. Nat. Rev. Genet. 2020, 21, 581–596. [Google Scholar] [CrossRef]
- Stupnikov, A.; Tripathi, S.; de Matos Simoes, R.; McArt, D.; Salto-Tellez, M.; Glazko, G.; Dehmer, M.; Emmert-Streib, F. samExploreR: Exploring reproducibility and robustness of RNA-seq results based on SAM files. Bioinformatics 2016, 32, 3345–3347. [Google Scholar] [CrossRef] [Green Version]
- Schurch, N.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberg, A.; Bot, B.; Grill, D.; Poland, G.; Therneau, T. Technical and biological variance structure in mRNA-Seq data: Life in the real world. BMC Genom. 2012, 13, 304. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Lee, E.; Kim, J. Analysis of Technical and Biological Variability in Single-Cell RNA Sequencing; Humana Press: Totowa, NJ, USA, 2019; Volume 1935, pp. 25–43. [Google Scholar] [CrossRef]
- Moutsopoulos, I.; Maischak, L.; Lauzikaite, E.; Vasquez Urbina, S.; Williams, E.; Drost, H.G.; Mohorianu, I. noisyR: Enhancing biological signal in sequencing datasets by characterizing random technical noise. Nucleic Acids Res. 2021, 49, e83. [Google Scholar] [CrossRef]
- Ma, X.; Shao, Y.; Tian, L.; Flasch, D.; Mulder, H.; Edmonson, M.; Liu, Y.; Chen, X.; Chen, X.; Newman, S.; et al. Analysis of error profiles in deep next-generation sequencing data. Genome Biol. 2019, 20, 50. [Google Scholar] [CrossRef] [Green Version]
- Ross, M.; Russ, C.; Costello, M.; Hollinger, A.; Lennon, N.; Hegarty, R.; Nusbaum, C.; Jaffe, D. Characterizing and measuring bias in sequence data. Genome Biol. 2013, 14, R51. [Google Scholar] [CrossRef] [Green Version]
- Sorefan, K.; Pais, H.; Hall, A.; Kozomara, A.; Griffiths-Jones, S.; Moulton, V.; Dalmay, T. Reducing ligation bias of small RNAs in libraries for next generation sequencing. Silence 2012, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Hicks, S.; Townes, F.; Teng, M.; Irizarry, R. Missing data and technical variability in single-cell RNA-sequencing experiments. Biostat 2017, 19, 562–578. [Google Scholar] [CrossRef]
- Reuter, J.; Spacek, D.; Snyder, M. High-Throughput Sequencing Technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Eraslan, G.; Simon, L.; Mircea, M.; Mueller, N.; Theis, F. Single-cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 2019, 10, 390. [Google Scholar] [CrossRef]
- Chazarra-Gil, R.; Dongen, S.; Kiselev, V.; Hemberg, M. Flexible comparison of batch correction methods for single-cell RNA-seq using BatchBench. Nucleic Acids Res. 2021, 49, e42. [Google Scholar] [CrossRef]
- Srivastava, A.; Malik, L.; Sarkar, H.; Zakeri, M.; Almodaresi, F.; Soneson, C.; Love, M.; Kingsford, C.; Patro, R. Alignment and mapping methodology influence transcript abundance estimation. Genome Biol. 2020, 21, 239. [Google Scholar] [CrossRef]
- Dillies, M.; Rau, A.; Aubert, J.; Hennequet-Antier, C.; Jeanmougin, M.; Servant, N.; Keime, C.; Marot, G.; Castel, D.; Estelle, J.; et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Briefings Bioinform. 2012, 14, 671–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mccarthy, D.; Chen, Y.; Smyth, G. Differential expression analysis of multifactor SRNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, V.; Natarajan, K.; Ly, L.H.; Miragaia, R.; Labalette, C.; Macaulay, I.; Cvejic, A.; Teichmann, S. Power analysis of single cell RNA-sequencing experiments. Nat. Methods 2017, 14, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Nakato, R.; Shirahige, K. Recent advances in ChIP-seq analysis: From quality management to whole-genome annotation. Briefings Bioinform. 2016, 18, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Chung, D.; Kuan, P.; Li, B.; Sanalkumar, R.; Liang, K.; Bresnick, E.; Dewey, C.; Keles, S. Discovering Transcription Factor Binding Sites in Highly Repetitive Regions of Genomes with Multi-Read Analysis of ChIP-Seq Data. PLoS Comput. Biol. 2011, 7, e1002111. [Google Scholar] [CrossRef] [PubMed]
- Dal Molin, A.; Camillo, B. How to design a single-cell RNA-sequencing experiment: Pitfalls, challenges and perspectives. Briefings Bioinform. 2018, 20, 1384–1394. [Google Scholar] [CrossRef]
- Goh, W.W.B.; Wang, W.; Wong, L. Why Batch Effects Matter in Omics Data, and How to Avoid Them. Trends Biotechnol. 2017, 35, 498–507. [Google Scholar] [CrossRef]
- Buttner, M.; Miao, Z.; Wolf, F.o. A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods 2019, 16, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.; Scharpf, R.; Bravo, H.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.; Ang, K.S.; Chevrier, M.; Zhang, X.; Lee, N.; Goh, M.; Chen, J. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020, 21, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Parmigiani, G.; Johnson, W.E. ComBat-seq: Batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020, 2, lqaa078. [Google Scholar] [CrossRef] [PubMed]
- Holmström, S.; Hautaniemi, S.; Häkkinen, A. POIBM: Batch correction of heterogeneous RNA-seq datasets through latent sample matching. Bioinformatics 2022, 38, 2474–2480. [Google Scholar] [CrossRef]
- Haghverdi, L.; Lun, A.; Morgan, M.; Marioni, J. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef]
- Lakkis, J.; Wang, D.; Zhang, Y.; Hu, G.; Wang, K.; Pan, H.; Ungar, L.; Reilly, M.; Li, X.; Li, M. A joint deep learning model enables simultaneous batch effect correction, denoising and clustering in single-cell transcriptomics. Genome Res. 2021, 31, 1753–1766. [Google Scholar] [CrossRef]
- Fei, T.; Yu, T. scBatch: Batch-effect correction of RNA-seq data through sample distance matrix adjustment. Bioinformatics 2020, 36, 3115–3123. [Google Scholar] [CrossRef]
- Fei, T.; Zhang, T.; Shi, W.; Yu, T. Mitigating the adverse impact of batch effects in sample pattern detection. Bioinformatics 2018, 34, 2634–2641. [Google Scholar] [CrossRef] [Green Version]
- Mohorianu, I.; Bretman, A.; Smith, D.; Fowler, E.; Dalmay, T.; Chapman, T. Genomic responses to socio-sexual environment in male Drosophila melanogaster exposed to conspecific rivals. RNA 2017, 23, 1048–1059. [Google Scholar] [CrossRef]
- Yang, P.; Humphrey, S.; Cinghu, S.; Pathania, R.; Oldfield, A.; Kumar, D.; Perera, D.; Yang, J.; James, D.; Mann, M.; et al. Multi-omic Profiling Reveals Dynamics of the Phased Progression of Pluripotency. Cell Syst. 2019, 8, 427–445.e10. [Google Scholar] [CrossRef] [Green Version]
- Cuomo, A.; Seaton, D.; McCarthy, D.; Martinez, I.; Bonder, M.; Garcia-Bernardo, J.; Amatya, S.; Madrigal, P.; Isaacson, A.; Buettner, F.; et al. Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat. Commun. 2020, 11, 810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mende, N.; Bastos, H.; Santoro, A.; Sham, K.; Mahbubani, K.; Curd, A.; Takizawa, H.; Wilson, N.; Göttgens, B.; Saeb-Parsy, K.; et al. Quantitative and molecular differences distinguish adult human medullary and extramedullary haematopoietic stem and progenitor cell landscapes. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Thurmond, J.; Goodman, J.; Strelets, V.; Attrill, H.; Gramates, L.; Marygold, S.; BB, M.; Millburn, G.; Antonazzo, G.; Trovisco, V.; et al. FlyBase 2.0: The next generation. Nucleic Acids Res. 2018, 47, D759–D765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, F.; Ryan, D.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Yates, A.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Ridwan Amode, M.; Armean, I.; Azov, A.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2019, 48, D682–D688. [Google Scholar] [CrossRef]
- Bolstad, B.; Irizarry, R.; Åstrand, M.; Speed, T. A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Variance and Bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.; Mccarthy, D.; Smyth, G. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.; Eeckhoute, J.; Johnson, D.; Bernstein, B.; Nusbaum, C.; Myers, R.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S.; Krueger, F.; Segonds-Pichon, A.; Biggins, L.; Krueger, C.; Montgomery, J. FastQC. Babraham Institute. 2012. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 November 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single cell data. Cell 2018, 177, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckers, M.; Mohorianu, I.; Stocks, M.; Applegate, C.; Dalmay, T.; Moulton, V. Comprehensive processing of high throughput small RNA sequencing data including quality checking, normalization and differential expression analysis using the UEA sRNA Workbench. RNA 2017, 23, 823–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.; Terry, J.; Belgrader, P.; Ryvkin, P.; Bent, Z.; Wilson, R.; Ziraldo, S.; Wheeler, T.; McDermott, G.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef] [Green Version]
- Waltman, L.; van Eck, N. A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B 2013, 86, 471. [Google Scholar] [CrossRef]
- Gates, A.; Wood, I.; Hetrick, W.; Ahn, Y.Y. Element-centric clustering comparison unifies overlaps and hierarchy. Sci. Rep. 2019, 9, 8574. [Google Scholar] [CrossRef]
- Shahsavari, A.; Munteanu, A.; Mohorianu, I. ClustAssess: Tools for Assessing the Robustness of Single-Cell Clustering. bioRxiv 2022. [Google Scholar] [CrossRef]
- Mohorianu, I.; Schwach, F.; Jing, R.; Lopez-Gomollon, S.; Moxon, S.; Szittya, G.; Sorefan, K.; Moulton, V.; Dalmay, T. Profiling of short RNAs during fleshy fruit development reveals stage-specific sRNAome expression patterns. Plant J. Cell Mol. Biol. 2011, 67, 232–246. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Case | N Cells | Donor | N Cells (Donor) | Time Point | N Cells (Time Point) | Cluster | N Cells (Cluster) |
|---|---|---|---|---|---|---|---|
| Case 1 | 105 | hayt | 105 | day2 | 105 | 0 | 105 |
| Case 2 | 106 | pahc | 106 | day3 | 106 | 1 | 66 |
| 4 | 40 | ||||||
| Case 3 | 168 | melw | 94 | day0 | 168 | 3 | 168 |
| qunz | 74 | ||||||
| Case 5 | 168 | hayt | 168 | day1 | 61 | 9 | 61 |
| day3 | 107 | 1 | 107 | ||||
| Case 6 | 95 | melw | 47 | day1 | 95 | 5 | 45 |
| vils | 48 | 6 | 50 | ||||
| Case 8 | 217 | melw | 95 | day0 | 95 | 3 | 95 |
| naah | 122 | day3 | 122 | 2 | 122 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, E.C.; Chazarra-Gil, R.; Shahsavari, A.; Mohorianu, I. The Sum of Two Halves May Be Different from the Whole—Effects of Splitting Sequencing Samples Across Lanes. Genes 2022, 13, 2265. https://doi.org/10.3390/genes13122265
Williams EC, Chazarra-Gil R, Shahsavari A, Mohorianu I. The Sum of Two Halves May Be Different from the Whole—Effects of Splitting Sequencing Samples Across Lanes. Genes. 2022; 13(12):2265. https://doi.org/10.3390/genes13122265
Chicago/Turabian StyleWilliams, Eleanor C., Ruben Chazarra-Gil, Arash Shahsavari, and Irina Mohorianu. 2022. "The Sum of Two Halves May Be Different from the Whole—Effects of Splitting Sequencing Samples Across Lanes" Genes 13, no. 12: 2265. https://doi.org/10.3390/genes13122265