
A Comparative Genomics Approach for Analysis of Complete Mitogenomes of Five Actinidiaceae Plants

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Genome Sequencing
2.2. Mitogenome Assembly and Annotation
2.3. Repeat Sequences and Chloroplast to Mitochondrion DNA Transformation
2.4. RNA Editing Predicting and Codon Usage
2.5. Substitution Rate Calculation and Phylogenetic Inference
3. Results
3.1. Mitogenome Assembly, Annotation and Gene Features
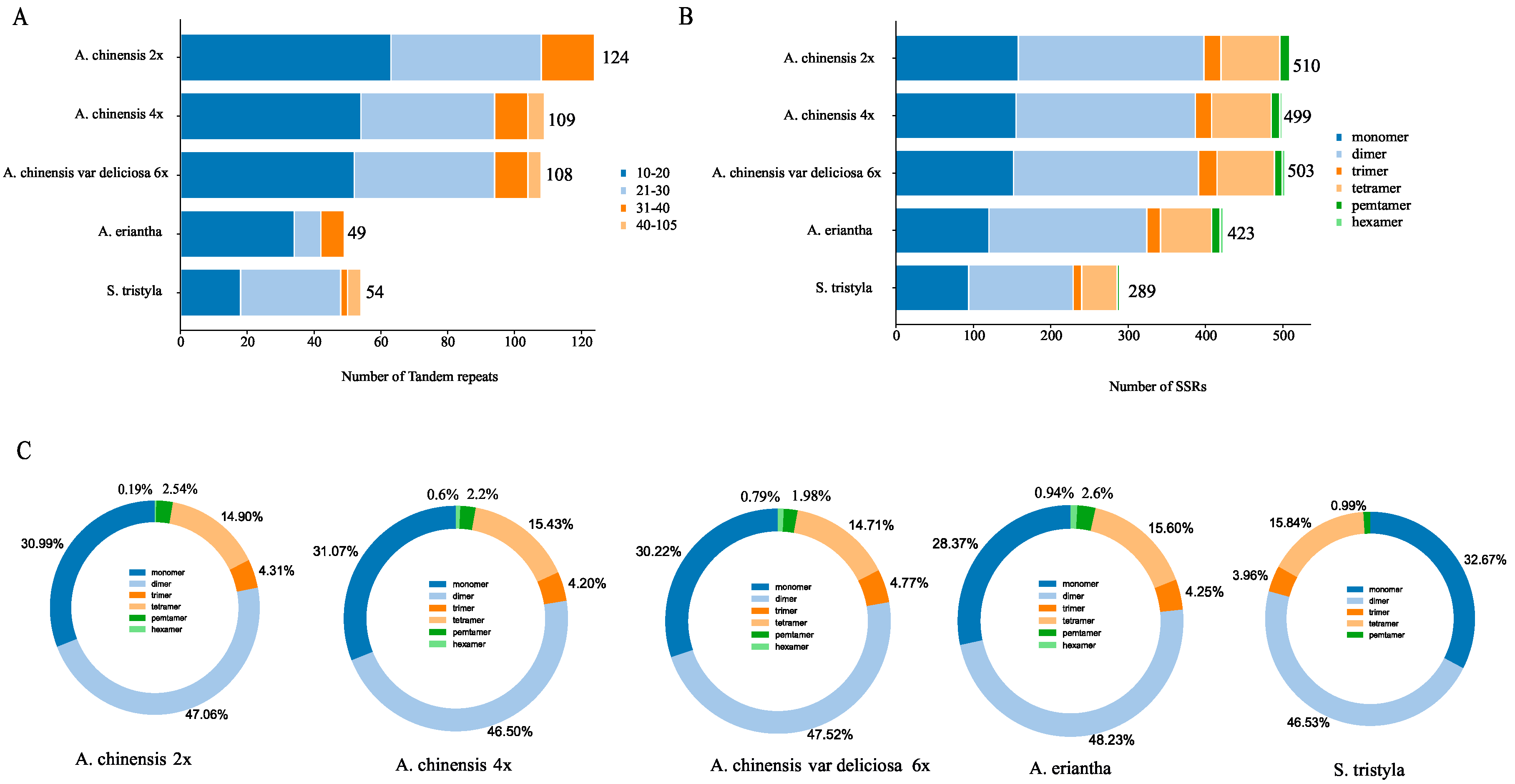
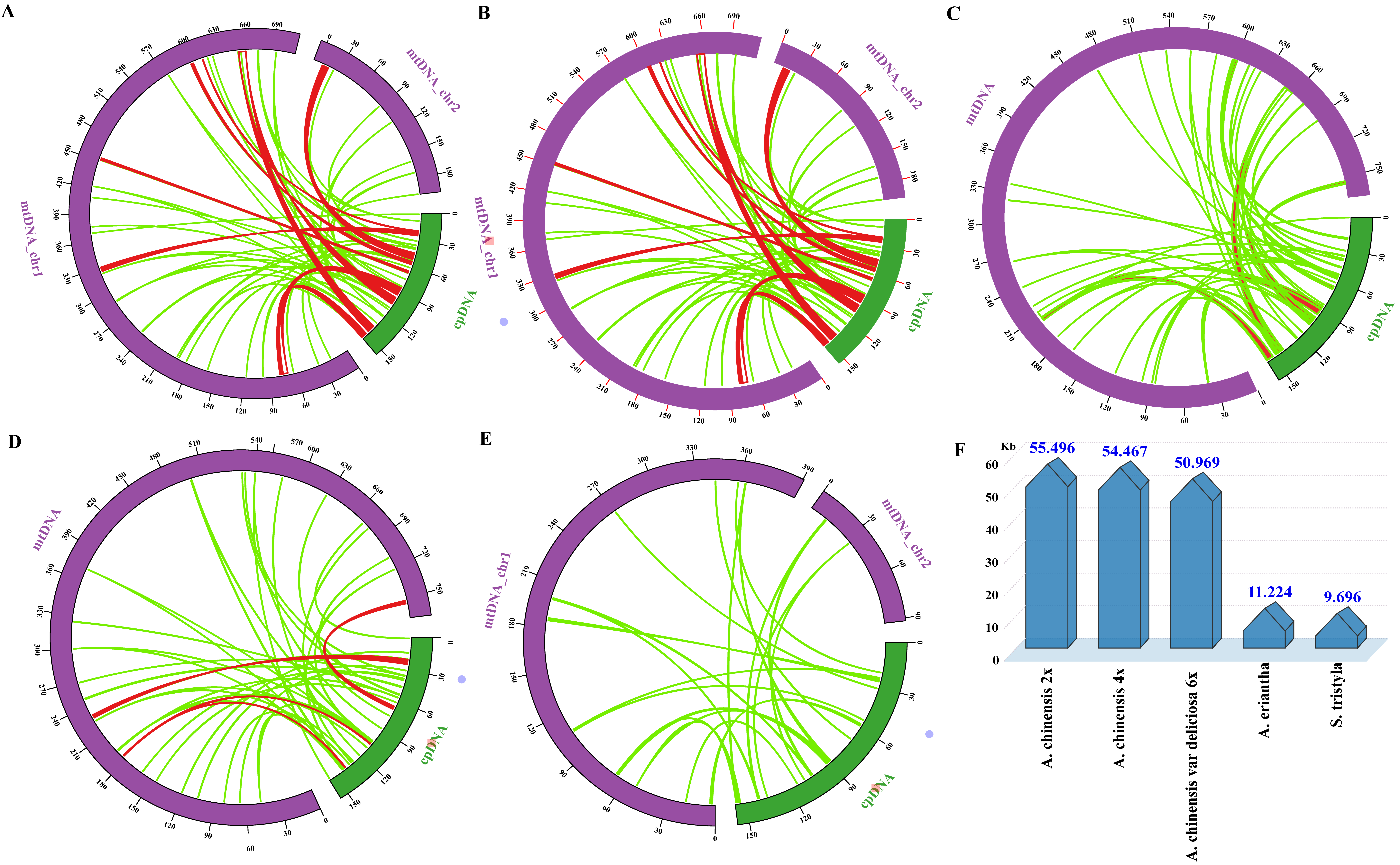
3.2. Repeat Sequences and Chloroplast-Derived Region Analysis
3.3. RNA Editing Sites and Codon Usage Analysis of PCGs
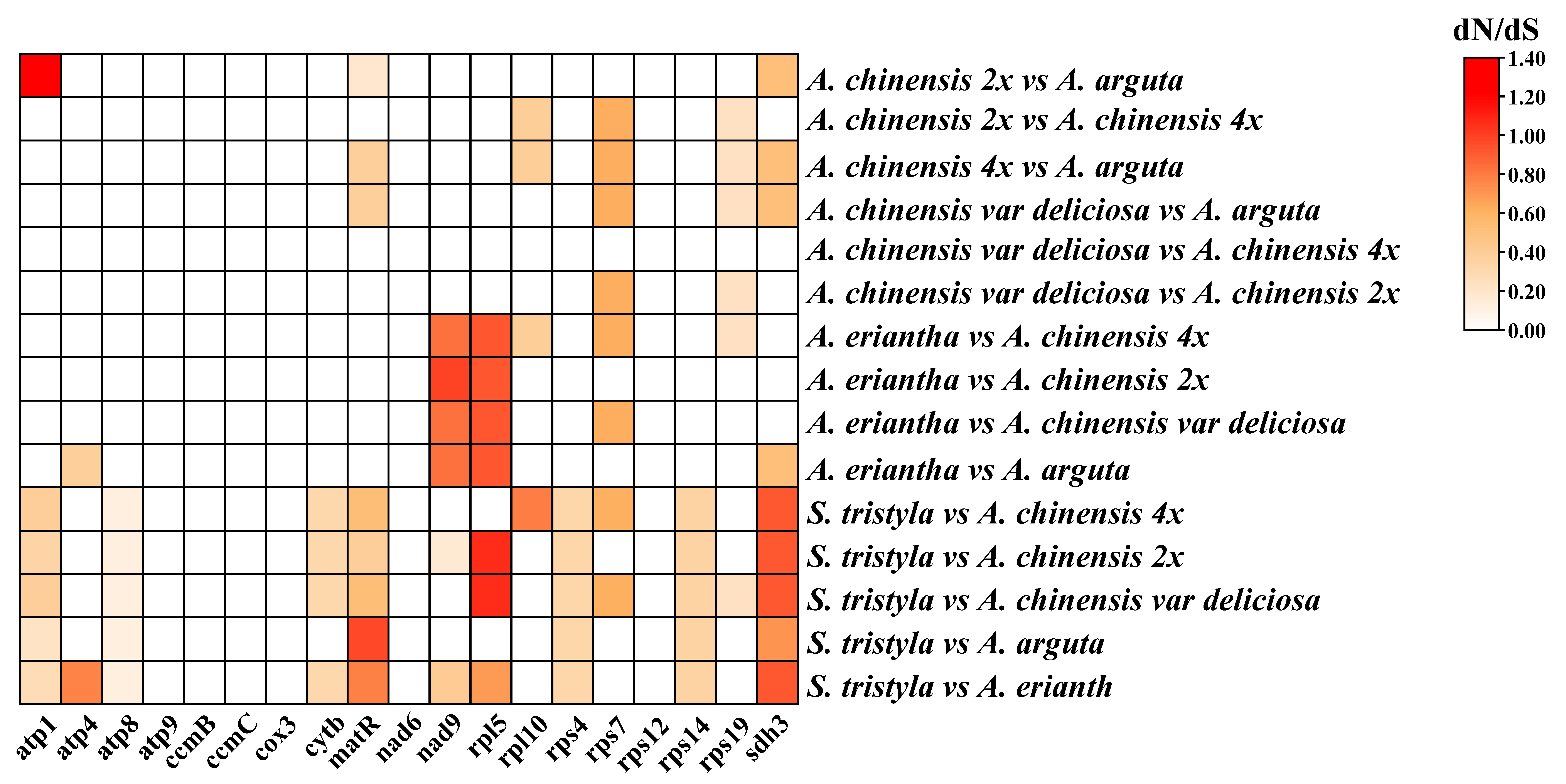
3.4. The Synonymous and Nonsynonymous Substitution Rate (Ka/Ks) and Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.W.; Lang, B.F.; Burger, G. Mitochondria of protists. Annu. Rev. Genet. 2004, 38, 477–524. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Wu, Z.; Sharbrough, J. Correction of persistent errors in Arabidopsis reference mitochondrial genomes. Plant Cell 2018, 30, 525–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, N.; Wang, X.; Li, J.; Bi, C.; Xu, Y.; Wu, D.; Ye, Q. Assembly and comparative analysis of complete mitochondrial genome sequence of an economic plant Salix suchowensis. Peer J. 2017, 5, e3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birky, C.W., Jr. Uniparental inheritance of mitochondrial and chloroplast genes: Mechanisms and evolution. Proc. Natl. Acad. Sci. USA 1995, 92, 11331–11338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.Q.; Liao, X.Z.; Zhang, X.N.; Tembrock, L.R.; Broz, A. Genomic architectural variation of plant mitochondria—A review of multichromosomal structuring. J. Syst. Evol. 2020, 60, 160–168. [Google Scholar] [CrossRef]
- Cole, L.W.; Guo, W.; Mower, J.P.; Palmer, J.D. High and variable rates of repeat-mediated mitochondrial genome rearrangement in a genus of plants. Mol. Biol. Evol. 2018, 35, 2773–2785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morley, S.A.; Nielsen, B.L. Plant mitochondrial DNA. Molecules 2017, 15, 17. [Google Scholar]
- Bergthorsson, U.; Adams, K.L.; Thomason, B.; Palmer, J.D. Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature 2003, 424, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xu, Y.; Shan, Y.; Pei, X.; Yong, S.; Liu, C.; Yu, J. Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta 2021, 254, 36. [Google Scholar] [CrossRef]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell 2011, 23, 2499–2513. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ni, Y.; Lin, Z.; Yang, L.; Chen, G.; Nijiati, N.; Hu, Y.; Chen, X. De novo assembly of the complete mitochondrial genome of sweet potato (Ipomoea batatas [L.] Lam) revealed the existence of homologous conformations generated by the repeat-mediated recombination. BMC Plant Biol. 2022, 22, 285. [Google Scholar] [CrossRef] [PubMed]
- Sang, S.F.; Mei, D.S.; Liu, J.; Zaman, Q.U.; Zhang, H.Y.; Hao, M.Y.; Fu, L.; Wang, H.; Cheng, H.F.; Hu, Q. Organelle genome composition and candidate gene identification for Nsa cytoplasmic male sterility in Brassica napus. BMC Genom. 2019, 20, 813. [Google Scholar] [CrossRef] [PubMed]
- Albert, V.A.; Jobson, R.W.; Michael, T.P.; Taylor, D.J. The carnivorous bladderwort (Utricularia, Lentibulariaceae): A system inflates. J. Exp. Bot. 2010, 61, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Phillips, W.S.; Brown, A.M.; Howe, D.K.; Peetz, A.B.; Blok, V.C.; Denver, D.R.; Zasada, I.A. The mitochondrial genome of Globodera ellingtonae is composed of two circles with segregated gene content and differential copy numbers. BMC Genom. 2016, 17, 706. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Li, W.; Gao, C.W.; Zhang, D.; Gao, L.Z. Deciphering tea tree chloroplast and mitochondrial genomes of Camellia sinensis var. assamica. Sci. Data 2019, 6, 209. [Google Scholar] [CrossRef] [Green Version]
- Richardson, A.O.; Rice, D.W.; Young, G.J.; Alverson, A.J.; Palmer, J.D. The “fossilized” mitochondrial genome of Liriodendron tulipifera: Ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 2013, 11, 29. [Google Scholar] [CrossRef] [Green Version]
- Bi, C.; Paterson, A.H.; Wang, X.; Xu, Y.; Wu, D.; Qu, Y.; Jiang, A.; Ye, Q.; Ye, N. Analysis of the Complete mitochondrial genome sequence of the diploid cotton Gossypium raimondii by comparative genomics approaches. BioMed Res. Int. 2016, 2016, 5040598. [Google Scholar] [CrossRef] [Green Version]
- Mwamuye, M.M.; Obara, I.; Elati, K.; Odongo, D.; Bakheit, M.A.; Jongejan, F. Unique mitochondrial single nucleotide polymorphisms demonstrate resolution potential to discriminate Theileria parva vaccine and buffalo-derived strains. Life 2020, 10, 334. [Google Scholar] [CrossRef]
- Zhang, D.; Xing, Y.; Xu, L.; Zhao, R.; Yang, Y.; Zhang, T.; Li, S.; Bao, G.; Ao, W.; Liu, T. Comparative analysis of the mitochondrial genome sequences of two medicinal plants: Arctium lappa and A. tomentosum; Research Square: Durham, NC, USA, 2020. [Google Scholar]
- Mower, J.P.; Sloan, D.B.; Alverson, A.J. Plant mitochondrial genome diversity: The genomics revolution. Plant Genome Divers. 2012, 1, 123–144. [Google Scholar]
- Dietrich, A.; Maréchal-Drouard, L.; Carneiro, V.; Cosset, A.; Small, I. A single base change prevents import of cytosolic tRNA(Ala) into mitochondria in transgenic plants. Plant J. 1996, 10, 913–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, R.; Bendahmane, A.; Renner, S.S. Sex chromosomes in land plants. Annu. Rev. Plant Biol. 2011, 62, 485–514. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, J.L. The scientific, economic, and social impacts of the New Zealand outbreak of bacterial canker of kiwifruit (Pseudomonas syringae pv. actinidiae). Annu. Rev. Phytopathol. 2017, 55, 377–399. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.M.; Fang, J.B.; Zhang, M.; Qi, X.J.; Lin, M.M.; Chen, J.Y. Molecular cloning and functional analysis of the NPR1 Homolog in kiwifruit (Actinidia eriantha). Front. Plant Sci. 2020, 11, 551201. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.L.; Wang, G.; Wang, T.; Jia, Z.H.; Guo, Z.R.; Zhang, J.Y. AdRAP2.3, a novel ethylene response factor VII from Actinidia deliciosa, enhances waterlogging resistance in transgenic tobacco through improving expression levels of PDC and ADH genes. Int. J. Mol. Sci. 2019, 20, 1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, L.M.; Swartzlander, D.; Meadows, K.L.; Wilkinson, K.D.; Corbett, A.H.; Doetsch, P.W. Dynamic compartmentalization of base excision repair proteins in response to nuclear and mitochondrial oxidative stress. Mol. Cell. Biol. 2009, 29, 794–807. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Ma, T.; Kang, M.; Ai, F.; Zhang, J.; Dong, G.; Liu, J. A high-quality Actinidia chinensis (kiwifruit) genome. Hortic. Res. 2019, 6, 117. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Ding, J.; Deng, D.; Tang, W.; Sun, H.; Liu, D.; Zhang, L.; Niu, X.; Zhang, X.; Meng, M.; et al. Draft genome of the kiwifruit Actinidia chinensis. Nat. Commun. 2013, 4, 2640. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Sun, X.; Yue, J.; Tang, X.; Jiao, C.; Yang, Y.; Niu, X.; Miao, M.; Zhang, D.; Huang, S.; et al. Chromosome-scale genome assembly of kiwifruit Actinidia eriantha with single-molecule sequencing and chromatin interaction mapping. GigaScience 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Pilkington, S.M.; Crowhurst, R.; Hilario, E.; Nardozza, S.; Fraser, L.; Peng, Y.; Gunaseelan, K.; Simpson, R.; Tahir, J.; Deroles, S.C.; et al. A manually annotated Actinidia chinensis var. chinensis (kiwifruit) genome highlights the challenges associated with draft genomes and gene prediction in plants. BMC Genom. 2018, 19, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, B.; Yang, Y.; Zhuang, Q.; Chen, S.; Liu, Y.; Huang, S. The comparative studies of complete chloroplast genomes in Actinidia (Actinidiaceae): Novel insights into heterogenous variation, clpP gene annotation and phylogenetic relationships. Mol. Genet. Genom. 2022, 297, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Emerman, A.B.; Bowman, S.K.; Barry, A.; Henig, N.; Patel, K.M.; Gardner, A.F.; Hendrickson, C.L. NEBNext direct: A novel, rapid, hybridization-based approach for the capture and library conversion of genomic regions of interest. Curr. Protoc. Mol. Biol. 2017, 119, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Istace, B.; Friedrich, A.; d’Agata, L.; Faye, S.; Payen, E.; Beluche, O.; Caradec, C.; Davidas, S.; Cruaud, C.; Liti, G.; et al. De novo assembly and population genomic survey of natural yeast isolates with the Oxford Nanopore MinION sequencer. GigaScience 2017, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef] [Green Version]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Ypung, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSe-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, w6–w11. [Google Scholar] [CrossRef] [Green Version]
- Misra, S.; Harris, N. Using Apollo to browse and edit genome annotations. Curr. Protoc. Bioinformatics 2006, 12, 9-5. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, w59–w64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, w253–w259. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing patforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res 2019, 47, w5–w10. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 2016, 44, w242–w245. [Google Scholar] [CrossRef]
- Wang, S.; Li, D.; Yao, X.; Song, Q.; Wang, Z.; Zhang, Q.; Zhong, C.; Liu, Y.; Huang, H. Evolution and diversification of kiwifruit mitogenomes through extensive whole-genome rearrangement and mosaic loss of intergenic sequences in a highly variable region. Genome Biol. Evol. 2019, 11, 1192–1206. [Google Scholar] [CrossRef]
- Rawal, H.C.; Kumar, P.M.; Bera, B.; Singh, N.K.; Mondal, T.K. Decoding and analysis of organelle genomes of Indian tea (Camellia assamica) for phylogenetic confirmation. Genomics 2020, 112, 659–668. [Google Scholar] [CrossRef]
- Zhao, N.; Wang, Y.; Hua, J. The roles of mitochondrion in intergenomic gene transfer in plants: A source and a pool. Int. J. Mol. Sci. 2018, 19, 547. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Garcia, L.; Rodriguez-Bonilla, L.; Rohde, J.; Smith, T.; Zalapa, J. Pacbio sequencing reveals identical organelle genomes between American cranberry (Vaccinium macrocarpon Ait.) and a wild relative. Genes 2019, 10, 291. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Grover, C.E.; Chen, Z.; Wendel, J.F.; Hua, J. Intergenomic gene transfer in diploid and allopolyploid Gossypium. BMC Plant Biol. 2019, 19, 492. [Google Scholar] [CrossRef] [Green Version]
- Lonsdale, D.M. A review of the structure and organization of the mitochondrial genome of higher plants. Plant Mol. Biol. 1984, 3, 201–206. [Google Scholar] [CrossRef]
- Xu, Y.; Cheng, W.; Xiong, C.; Jiang, X.; Wu, K.; Gong, B. Genetic diversity and association analysis among germplasms of diospyros kaki in Zhejiang province Based on SSR Markers. Forests 2021, 12, 422. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, M.; Chen, X.; Liu, Y.; Liu, B.; Li, J.; Wang, R.; Zhao, K.; Wu, J. Rearrangement and domestication as drivers of Rosaceae mitogenome plasticity. BMC Biol. 2022, 20, 181. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Dong, Y.; Cheng, W.; Wu, K.; Gao, H.; Liu, L.; Xu, L.; Gong, B. Characterization and phylogenetic analysis of the complete mitochondrial genome sequence of Diospyros oleifera, the first representative from the family Ebenaceae. Heliyon 2022, 8, e09870. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Grewe, F.; Fan, W.; Young, G.J.; Knoop, V.; Palmer, J.D.; Mower, J.P. Ginkgo and Welwitschia mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol. Biol. Evol. 2016, 33, 1448–1460. [Google Scholar] [CrossRef] [Green Version]
- Orton, L.M.; Fitzek, E.; Feng, X.; Grayburn, W.S.; Mower, J.P.; Liu, K.; Zhang, C.; Duvall, M.R.; Yin, Y. Zygnema circumcarinatum UTEX 1559 chloroplast and mitochondrial genomes provide insight into land plant evolution. J. Exp. Bot. 2020, 71, 3361–3373. [Google Scholar] [CrossRef]
- Xue, J.Y.; Wang, Y.; Chen, M.; Dong, S.; Shao, Z.Q.; Liu, Y. Maternal Inheritance of U’s Triangle and Evolutionary Process of Brassica Mitochondrial Genomes. Front. Plant Sci. 2020, 11, 805. [Google Scholar]
- Cheng, Y.; He, X.; Priyadarshani, S.V.G.N.; Wang, Y.; Ye, L.; Shi, C.; Ye, K.; Zhou, Q.; Luo, Z.; Deng, F.; et al. Assembly and comparative analysis of the complete mitochondrial genome of Suaeda glauca. BMC Genom. 2021, 22, 167. [Google Scholar] [CrossRef]
- Niu, Y.; Gao, C.; Liu, J. Complete mitochondrial genomes of three Mangifera species, their genomic structure and gene transfer from chloroplast genomes. BMC Genom. 2022, 23, 147. [Google Scholar] [CrossRef]
- Adams, K.L.; Qiu, Y.L.; Stoutemyer, M.; Palmer, J.D. Punctuated evolution of mitochondrial gene content: High and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. USA 2002, 99, 9905–9912. [Google Scholar] [CrossRef] [Green Version]
- Pinard, D.; Myburg, A.A.; Mizrachi, E. The plastid and mitochondrial genomes of Eucalyptus grandis. BMC Genom. 2019, 20, 132. [Google Scholar] [CrossRef]
- Lukeš, J.; Kaur, B.; Speijer, D. RNA Editing in Mitochondria and Plastids: Weird and Widespread. Trends Genet. 2021, 37, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Small, I.D.; Schallenberg-Rüdinger, M.; Takenaka, M.; Mireau, H.; Ostersetzer-Biran, O. Plant organellar RNA editing: What 30 years of research has revealed. Plant J. 2020, 101, 1040–1056. [Google Scholar] [CrossRef] [PubMed]
- Monnens, M.; Thijs, S.; Briscoe, A.G.; Clark, M.; Frost, E.J.; Littlewood, D.T.J.; Sewell, M.; Smeets, K.; Artois, T.; Vanhove, M.P.M. The first mitochondrial genomes of endosymbiotic rhabdocoels illustrate evolutionary relaxation of atp8 and genome plasticity in flatworms. Int. J. Biol. Macromol. 2020, 162, 454–469. [Google Scholar] [CrossRef] [PubMed]
- Gerke, P.; Szövényi, P.; Neubauer, A.; Lenz, H.; Gutmann, B.; McDowell, R.; Small, I.; Schallenberg-Rüdinger, M.; Knoop, V. Towards a plant model for enigmatic U-to-C RNA editing: The organelle genomes, transcriptomes, editomes and candidate RNA editing factors in the hornwort Anthoceros agrestis. New Phytol 2020, 225, 1974–1992. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, X. Analysis of synonymous codon usage patterns in different plant mitochondrial genomes. Mol. Biol. Rep. 2009, 36, 2039–2046. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, Z.; Vang, S.; Yu, J.; Wong, G.K.; Wang, J. Correlation between Ka/Ks and Ks is related to substitution model and evolutionary lineage. J. Mol. Evol. 2009, 68, 414–423. [Google Scholar] [CrossRef]
- Yu, X.; Duan, Z.; Wang, Y.; Zhang, Q.; Li, W. Sequence analysis of the complete mitochondrial genome of a medicinal Plant, vitex rotundifolia Linnaeus f. (Lamiales: Lamiaceae). Genes 2022, 13, 839. [Google Scholar] [CrossRef]
- Wang, W.C.; Chen, S.Y.; Zhang, X.Z. Chloroplast genome evolution in Actinidiaceae: clpP loss, heterogenous divergence and phylogenomic practice. PLoS ONE 2016, 11, e0162324. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Ling, C.; Zhang, H.; Hussain, Q.; Lyu, S.; Zheng, G.; Liu, Y. A Comparative Genomics Approach for Analysis of Complete Mitogenomes of Five Actinidiaceae Plants. Genes 2022, 13, 1827. https://doi.org/10.3390/genes13101827
Yang J, Ling C, Zhang H, Hussain Q, Lyu S, Zheng G, Liu Y. A Comparative Genomics Approach for Analysis of Complete Mitogenomes of Five Actinidiaceae Plants. Genes. 2022; 13(10):1827. https://doi.org/10.3390/genes13101827
Chicago/Turabian StyleYang, Jun, Chengcheng Ling, Huamin Zhang, Quaid Hussain, Shiheng Lyu, Guohua Zheng, and Yongsheng Liu. 2022. "A Comparative Genomics Approach for Analysis of Complete Mitogenomes of Five Actinidiaceae Plants" Genes 13, no. 10: 1827. https://doi.org/10.3390/genes13101827