
Combining Gene Mutation with Expression of Candidate Genes to Improve Diagnosis of Escobar Syndrome
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Data Collection
2.2. DNA Extraction
2.3. Sanger Sequencing
2.4. Whole Exome Sequencing and Bioinformatics Analysis
2.5. RNA Extraction and Real-Time Quantitative PCR
2.6. Statistical Analysis
3. Results
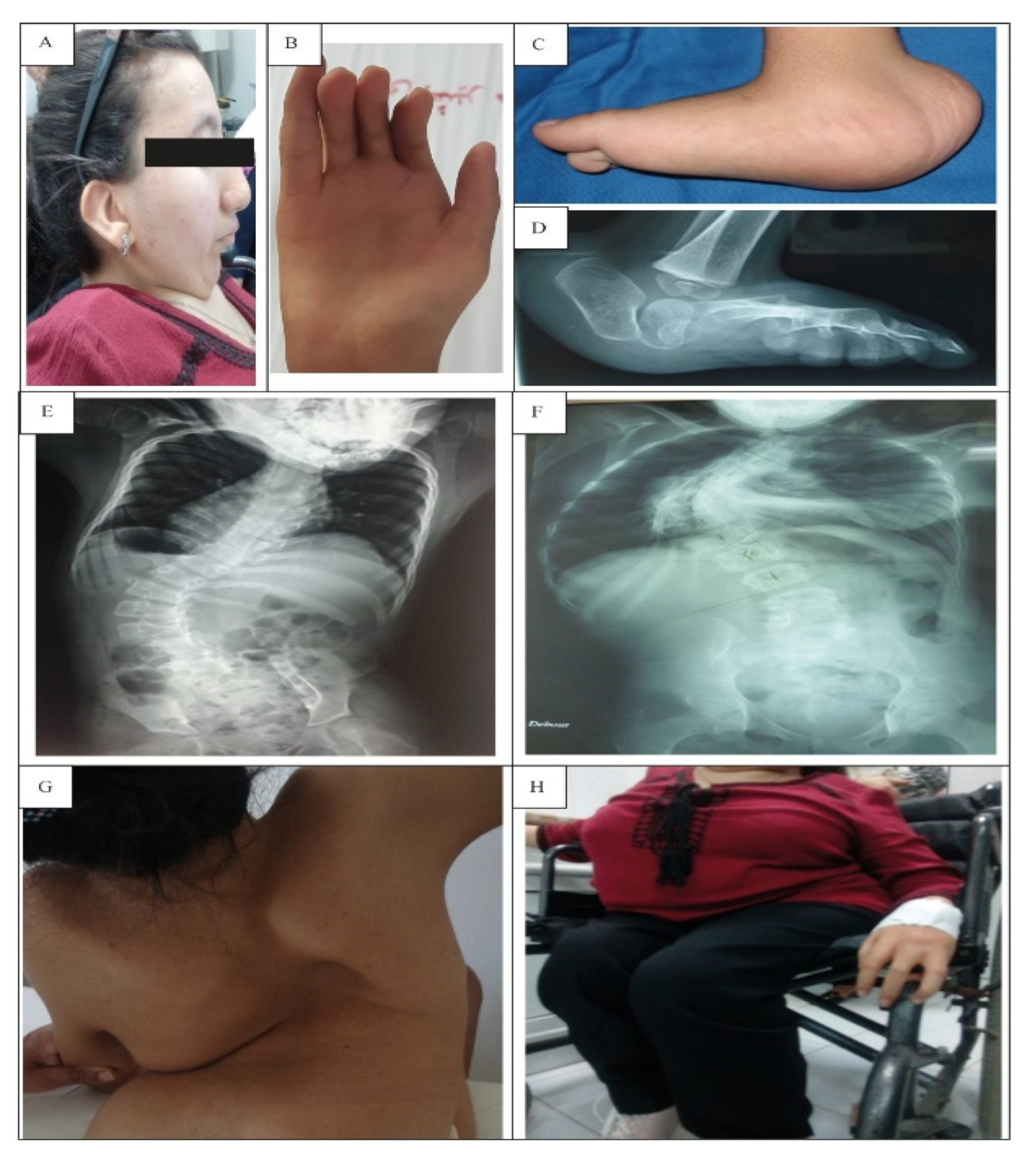
3.1. Clinical Investigation
3.1.1. Prenatal Abnormalities
3.1.2. Facial and Skin Anomalies
3.1.3. Skeletal Deformities
3.1.4. Genealogical Data of the Patients
3.1.5. History of LMPS Cases
3.2. Genetic Findings
3.2.1. Patient 4 and 5
3.2.2. Patient 1
3.2.3. Patients 2 and 3
3.3. Genetic Particularity of Patient 3
Oligogenic Inheritance
3.4. Decreased IGF1 Expression in Escobar Patients with Short Stature
3.5. Alteration of POLG1 Expression in Escobar Patients and Its Possible Link with TPM2 Mutation
4. Discussion
4.1. Clinical and Instrumental Findings
4.2. Molecular Diagnosis
4.2.1. Genetic Investigation of genes Responsible for Escobar Syndrome
4.2.2. Investigation of the Clinical Particularities in Patient 3
4.3. Cases of LMPS in Our Cohort
4.4. Investigation of Candidate Genes’ Expression Associated with Escobar Syndrome: IGF-1 and POLG1
4.4.1. IGF-1 Expression
4.4.2. POLG1 Expression
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beecroft, S.J.; Lombard, M.; Mowat, D.; McLean, C.; Cairns, A.; Davis, M.; Laing, N.; Ravenscroft, G. Genetics of neuromuscular fetal akinesia in the genomics era. J. Med. Genet. 2018, 55, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Escobar, V.; Bixler, D.; Gleiser, S.; Weaver, D.D.; Gibbs, T. Multiple pterygium syndrome. Am. J. Dis. Child. 1978, 132, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Müller, J.S.; Stricker, S.; Megarbane, A.; Rajab, A.; Lindner, T.H.; Cohen, M.; Chouery, E.; Adaimy, L.; Ghanem, I.; et al. Escobar Syndrome Is a Prenatal Myasthenia Caused by Disruption of the Acetylcholine Receptor Fetal γ Subunit. Am. J. Hum. Genet. 2006, 79, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Morgan, N.; Brueton, L.A.; Cox, P.; Greally, M.T.; Tolmie, J.; Pasha, S.; Aligianis, I.A.; van Bokhoven, J.; Marton, T.; Al-Gazali, L.; et al. Mutations in the Embryonal Subunit of the Acetylcholine Receptor (CHRNG) Cause Lethal and Escobar Variants of Multiple Pterygium Syndrome. Am. J. Hum. Genet. 2006, 79, 390–395. [Google Scholar] [CrossRef]
- Mishina, M.; Takai, T.; Imoto, K.; Noda, M.; Takahashi, T.; Numa, S.; Methfessel, C.; Sakmann, B. Molecular distinction between fetal and adult forms of muscle acetylcholine receptor. Nature 1986, 321, 406–411. [Google Scholar] [CrossRef]
- Vogt, J.; Morgan, N.V.; Rehal, P.; Faivre, L.; Brueton, L.A.; Becker, K.; Fryns, J.P.; Holder, S.; Islam, L.; Kivuva, E.; et al. CHRNG genotype–phenotype correlations in the multiple pterygium syndromes. J. Med. Genet. 2012, 49, 21–26. [Google Scholar] [CrossRef]
- Kariminejad, A.; Almadani, N.; Khoshaeen, A.; Olsson, B.; Moslemi, A.-R.; Tajsharghi, H. Truncating CHRNG mutations associated with interfamilial variability of the severity of the Escobar variant of multiple pterygium syndrome. BMC Genet. 2016, 17, 71. [Google Scholar] [CrossRef]
- Hesselmans, L.F.G.M.; Jennekens, F.G.I.; Oord, C.J.M.V.D.; Veldman, H.; Vincent, A. Development of innervation of skeletal muscle fibers in man: Relation to acetylcholine receptors. Anat. Rec. 1993, 236, 553–562. [Google Scholar] [CrossRef]
- Missias, A.C.; Chu, G.C.; Klocke, B.J.; Sanes, J.R.; Merlie, J.P. Maturation of the Acetylcholine Receptor in Skeletal Muscle: Regulation of the AChR γ-to-ϵ Switch. Dev. Biol. 1996, 179, 223–238. [Google Scholar] [CrossRef]
- Yumoto, N.; Wakatsuki, S.; Sehara-Fujisawa, A. The acetylcholine receptor γ-to-ε switch occurs in individual endplates. Biochem. Biophys. Res. Commun. 2005, 331, 1522–1527. [Google Scholar] [CrossRef]
- Carrera-García, L.; Natera-de Benito, D.; Dieterich, K.; de la Banda, M.G.; Felter, A.; Inarejos, E.; Codina, A.; Jou, C.; Roldan, M.; Palau, F.; et al. CHRNG-related nonlethal multiple pterygium syndrome: Muscle imaging pattern and clinical, histopathological, and molecular genetic findings. Am. J. Med. Genet. Part A 2019, 179, 915–926. [Google Scholar] [CrossRef]
- Robinson, K.G.; Viereck, M.J.; Margiotta, M.V.; Gripp, K.W.; Abdul-Rahman, O.A.; Akins, R.E. Neuromotor synapses in Escobar syndrome. Am. J. Med. Genet. Part A 2013, 161, 3042–3048. [Google Scholar] [CrossRef]
- Al Kaissi, A.; Ryabykh, S.; Ochirova, P.; Bouchoucha, S.; Kenis, V.; Shboul, M.; Ganger, R.; Grill, F.; Kircher, S.G. Arthrogryposis is a descriptive term, not a specific disease entity: Escobar syndrome is an Example. Minerva Pediatr. 2020. online ahead of print. [Google Scholar] [CrossRef]
- Sandweiss, A.J.; Patel, S.; Bader, M.Y.; Kylat, R.I. A Truncating Variant of CHRNG as a Cause of Escobar Syndrome: A Multiple Pterygium Syndrome Subtype. J. Pediatr. Genet. 2020, 11, 144–146. [Google Scholar] [CrossRef]
- Al Kaissi, A.; Kenis, V.; Laptiev, S.; Ghachem, M.B.; Klaushofer, K.; Ganger, R.; Grill, F. Is webbing (pterygia) a constant feature in patients with Escobar syndrome? Orthop. Surg. 2013, 5, 297–301. [Google Scholar] [CrossRef]
- Vogt, J.; Harrison, B.J.; Spearman, H.; Cossins, J.; Vermeer, S.; Cate, L.N.T.; Morgan, N.V.; Beeson, D.; Maher, E.R. Mutation Analysis of CHRNA1, CHRNB1, CHRND, and RAPSN Genes in Multiple Pterygium Syndrome/Fetal Akinesia Patients. Am. J. Hum. Genet. 2008, 82, 222–227. [Google Scholar] [CrossRef]
- Monnier, N.; Lunardi, J.; Marty, I.; Mezin, P.; Labarre-Vila, A.; Dieterich, K.; Jouk, P.S. Absence of β-tropomyosin is a new cause of Escobar syndrome associated with nemaline myopathy. Neuromuscul. Disord. 2009, 19, 118–123. [Google Scholar] [CrossRef]
- Schirwani, S.; Sarkozy, A.; Phadke, R.; Childs, A.-M.; Mein, R.; Ismail, A.; Smith, A.; Muntoni, F.; Hobson, E.; Pysden, K. Homozygous intronic variants in TPM2 cause recessively inherited Escobar variant of multiple pterygium syndrome and congenital myopathy. Neuromuscul. Disord. 2021, 31, 359–366. [Google Scholar] [CrossRef]
- Vogt, J.; Al-Saedi, A.; Willis, T.; Male, A.; McKie, A.; Kiely, N.; Maher, E.R. A recurrent pathogenic variant in TPM2 reveals further phenotypic and genetic heterogeneity in multiple pterygium syndrome-related disorders. Clin. Genet. 2020, 97, 908–914. [Google Scholar] [CrossRef]
- Pollazzon, M.; Caraffi, S.G.; Faccioli, S.; Rosato, S.; Fodstad, H.; Campos-Xavier, B.; Soncini, E.; Comitini, G.; Frattini, D.; Grimaldi, T.; et al. Clinical and Genetic Findings in a Series of Eight Families with Arthrogryposis. Genes 2021, 13, 29. [Google Scholar] [CrossRef]
- Filges, I.; Hall, J.G. Failure to identify antenatal multiple congenital contractures and fetal akinesia—Proposal of guidelines to improve diagnosis. Prenat. Diagn. 2013, 33, 61–74. [Google Scholar] [CrossRef]
- Stals, K.L.; Wakeling, M.; Baptista, J.; Caswell, R.; Parrish, A.; Rankin, J.; Tysoe, C.; Jones, G.; Gunning, A.C.; Lango Allen, H.; et al. Diagnosis of lethal or prenatal-onset autosomal recessive disorders by parental exome sequencing. Prenat. Diagn. 2018, 38, 33–43. [Google Scholar] [CrossRef]
- Bikle, D.D.; Tahimic, C.; Chang, W.; Wang, Y.; Philippou, A.; Barton, E.R. Role of IGF-I signaling in muscle bone interactions. Bone 2015, 80, 79–88. [Google Scholar] [CrossRef]
- LeComte, M.-J.; Bertolus, C.; Ramanantsoa, N.; Saurini, F.; Callebert, J.; Sénamaud-Beaufort, C.; Ringot, M.; Bourgeois, T.; Matrot, B.; Collet, C.; et al. Acetylcholine Modulates the Hormones of the Growth Hormone/Insulinlike Growth Factor-1 Axis during Development in Mice. Endocrinology 2018, 159, 1844–1859. [Google Scholar] [CrossRef]
- Baron, J.; Sävendahl, L.; De Luca, F.; Dauber, A.; Phillip, M.; Wit, J.M.; Nilsson, O. Short and tall stature: A new paradigm emerges. Nat. Rev. Endocrinol. 2015, 11, 735–746. [Google Scholar] [CrossRef]
- Lamont, P.J.; Thorburn, D.R.; Fabian, V.; Vajsar, J.; Hawkins, C.; (Reisch), A.S.; Durling, H.; Laing, N.G.; Nevo, Y. Nemaline Rods and Complex I Deficiency in Three Infants with Hypotonia, Motor Delay and Failure to Thrive. Neuropediatrics 2004, 35, 302–306. [Google Scholar] [CrossRef]
- Mokbel, N.; Ilkovski, B.; Kreissl, M.; Memo, M.; Jeffries, C.M.; Marttila, M.; Lehtokari, V.-L.; Lemola, E.; Grönholm, M.; Yang, N.; et al. K7del is a common TPM2 gene mutation associated with nemaline myopathy and raised myofibre calcium sensitivity. Brain 2013, 136, 494–507. [Google Scholar] [CrossRef]
- Pula, S.; Urankar, K.; Norman, A.; Pierre, G.; Langton-Hewer, S.; Selby, V.; Mason, F.; Vijayakumar, K.; McFarland, R.; Taylor, R.W.; et al. A novel de novo ACTA1 variant in a patient with nemaline myopathy and mitochondrial Complex I deficiency. Neuromuscul. Disord. 2019, 30, 159–164. [Google Scholar] [CrossRef]
- Rahman, S.; Copeland, W.C. POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 2018, 15, 40–52. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Renaux, A.; Papadimitriou, S.; Versbraegen, N.; Nachtegael, C.; Boutry, S.; Nowé, A.; Smits, G.; Lenaerts, T. ORVAL: A novel platform for the prediction and exploration of disease-causing oligogenic variant combinations. Nucleic Acids Res. 2019, 47, W93–W98. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.; Van Heest, A.E.; Pleasure, D. Arthrogryposis: A Review and Update. J. Bone Jt. Surg. 2009, 91, 40–46. [Google Scholar] [CrossRef]
- Ferguson, J.; Wainwright, A. Arthrogryposis. Orthop. Trauma 2013, 27, 171–180. [Google Scholar] [CrossRef]
- DeMyer, W.; Baird, I. Mortality and skeletal malformations from amniocentesis and oligohydramnios in rats: Cleft palate, clubfoot, microstomia, and adactyly. Teratology 1969, 2, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.B.; Sokoloff, L. The role of movement in embryonic joint development. Dev. Biol. 1966, 14, 401–420. [Google Scholar] [CrossRef]
- Parashar, S.Y.; Anderson, P.J.; McLean, N.; David, D.J.; Djohansjah, M. Spectrum of Features in Pterygium Syndrome. Asian J. Surg. 2006, 29, 104–108. [Google Scholar] [CrossRef]
- Kodaganur, S.G.; Tontanahal, S.J.; Sarda, A.; Shah, M.H.; Bhat, V.; Kumar, A. Clinical phenotype and the lack of mutations in the CHRNG, CHRND, and CHRNA1 genes in two Indian families with Escobar syndrome. Clin. Dysmorphol. 2013, 22, 54–58. [Google Scholar] [CrossRef]
- Dahan-Oliel, N.; Dieterich, K.; Rauch, F.; Bardai, G.; Blondell, T.; Gustafson, A.; Hamdy, R.; Latypova, X.; Shazand, K.; Giampietro, P.; et al. The Clinical and Genotypic Spectrum of Scoliosis in Multiple Pterygium Syndrome: A Case Series on 12 Children. Genes 2021, 12, 1220. [Google Scholar] [CrossRef]
- Ochala, J. Thin filament proteins mutations associated with skeletal myopathies: Defective regulation of muscle contraction. Klin. Wochenschr. 2008, 86, 1197–1204. [Google Scholar] [CrossRef]
- Sung, S.S.; Brassington, A.-M.E.; Grannatt, K.; Rutherford, A.; Whitby, F.G.; Krakowiak, P.A.; Jorde, L.B.; Carey, J.C.; Bamshad, M. Mutations in Genes Encoding Fast-Twitch Contractile Proteins Cause Distal Arthrogryposis Syndromes. Am. J. Hum. Genet. 2003, 72, 681–690. [Google Scholar] [CrossRef]
- Donner, K.; Ollikainen, M.; Ridanpää, M.; Christen, H.J.; Goebel, H.H.; de Visser, M.; Pelin, K.; Wallgren-Pettersson, C. Mutations in the β-tropomyosin (TPM2) gene–a rare cause of nemaline myopathy. Neuromuscul. Disord. 2002, 12, 151–158. [Google Scholar] [CrossRef]
- Lehtokari, V.-L.; Groote, C.C.-D.; de Jonghe, P.; Marttila, M.; Laing, N.G.; Pelin, K.; Wallgren-Pettersson, C. Cap disease caused by heterozygous deletion of the β-tropomyosin gene TPM2. Neuromuscul. Disord. 2007, 17, 433–442. [Google Scholar] [CrossRef]
- Brandis, A.; Aronica, E.; Goebel, H.H. TPM2 mutation. Neuromuscul. Disord. 2008, 18, 1005. [Google Scholar] [CrossRef]
- Ben Arab, S.; Masmoudi, S.; Beltaief, N.; Hachicha, S.; Ayadi, H. Consanguinity and endogamy in Northern Tunisia and its impact on non-syndromic deafness. Genet. Epidemiol. Off. Publ. Int. Genet. Epidemiol. Soc. 2004, 27, 74–79. [Google Scholar] [CrossRef]
- North, K.N.; Laing, N.G.; Wallgren-Pettersson, C. Nemaline myopathy: Current concepts. The ENMC International Consortium and Nemaline Myopathy. J. Med. Genet. 1997, 34, 705–713. [Google Scholar] [CrossRef]
- Cassandrini, D.; Rodolico, C.; Trovato, R.; Rubegni, A.; Lenzi, S.; Fiorillo, C.; Baldacci, J.; Minetti, C.; Astrea, G.; Bruno, C.; et al. Congenital myopathies: Clinical phenotypes and new diagnostic tools. Ital. J. Pediatr. 2017, 43, 1–16. [Google Scholar] [CrossRef]
- Tajsharghi, H.; Kimber, E.; Holmgren, D.; Tulinius, M.; Oldfors, A. Distal arthrogryposis and muscle weakness associated with a β-tropomyosin mutation. Neurology 2007, 68, 772–775. [Google Scholar] [CrossRef]
- Gupta, V.A.; Beggs, A.H. Kelch proteins: Emerging roles in skeletal muscle development and diseases. Skelet. Muscle 2014, 4, 11. [Google Scholar] [CrossRef]
- Zhang, J. Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013; pp. 926–927. [Google Scholar]
- Chaillou, T.; Jackson, J.R.; England, J.H.; Kirby, T.J.; Richards-White, J.; Esser, K.A.; Dupont-Versteegden, E.E.; McCarthy, J.J. Identification of a conserved set of upregulated genes in mouse skeletal muscle hypertrophy and regrowth. J. Appl. Physiol. 2015, 118, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tingart, M.; Lecouturier, S.; Li, J.; Eschweiler, J. Identification of co-expression network correlated with different periods of adipogenic and osteogenic differentiation of BMSCs by weighted gene co-expression network analysis (WGCNA). BMC Genom. 2021, 22, 254. [Google Scholar] [CrossRef]
- Ravenscroft, G.; Miyatake, S.; Lehtokari, V.-L.; Todd, E.J.; Vornanen, P.; Yau, K.S.; Hayashi, Y.K.; Miyake, N.; Tsurusaki, Y.; Doi, H.; et al. Mutations in KLHL40 Are a Frequent Cause of Severe Autosomal-Recessive Nemaline Myopathy. Am. J. Hum. Genet. 2013, 93, 6–18. [Google Scholar] [CrossRef]
- Bowlin, K.M.; Embree, L.J.; Garry, M.G.; Garry, D.J.; Shi, X. Kbtbd5 is regulated by MyoD and restricted to the myogenic lineage. Differentiation 2013, 86, 184–191. [Google Scholar] [CrossRef]
- Garg, A.; O’Rourke, J.; Long, C.; Doering, J.; Ravenscroft, G.; Bezprozvannaya, S.; Nelson, B.R.; Beetz, N.; Li, L.; Chen, S.; et al. KLHL40 deficiency destabilizes thin filament proteins and promotes nemaline myopathy. J. Clin. Investig. 2014, 124, 3529–3539. [Google Scholar] [CrossRef]
- Schartner, V.; Romero, N.B.; Donkervoort, S.; Treves, S.; Munot, P.; Pierson, T.M.; Dabaj, I.; Malfatti, E.; Zaharieva, I.T.; Zorzato, F.; et al. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. 2016, 133, 517–533. [Google Scholar] [CrossRef]
- McArdle, P.F.; Rutherford, S.; Mitchell, B.D.; Damcott, C.M.; Wang, Y.; Ramachandran, V.; Ott, S.; Chang, Y.-P.C.; Levy, D.; Steinle, N. Nicotinic acetylcholine receptor subunit variants are associated with blood pressure; findings in the Old Order Amish and replication in the Framingham Heart Study. BMC Med. Genet. 2008, 9, 10–67. [Google Scholar] [CrossRef]
- Hall, J.G. Arthrogryposis (multiple congenital contractures): Diagnostic approach to etiology, classification, genetics, and general principles. Eur. J. Med. Genet. 2014, 57, 464–472. [Google Scholar] [CrossRef]
- Haliloglu, G.; Topaloglu, H. Arthrogryposis and fetal hypomobility syndrome. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 113, pp. 1311–1319. [Google Scholar] [CrossRef]
- Bayram, Y.; Karaca, E.; Akdemir, Z.C.; Yilmaz, E.O.; Tayfun, G.A.; Aydin, H.; Torun, D.; Bozdogan, S.T.; Gezdirici, A.; Isikay, S.; et al. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J. Clin. Investig. 2016, 126, 762–778. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, Z.; Zhao, T.; Wang, H.; Zhao, D.; Zhang, J.; Wang, Y.; Ding, Y.; Qiu, G. Adolescent Idiopathic Scoliosis and the Single-nucleotide Polymorphism of the Growth Hormone Receptor and IGF-1 Genes. Orthopedics 2009, 32, 411. [Google Scholar] [CrossRef]
- Moore, B.; Whitehead, A.; Davies, K. Short Stature, Growth Hormone Deficiency, and Primary IGF-1 Deficiency. In Advanced Practice in Endocrinology Nursing; Llahana, S., Follin, C., Yedinak, C., Grossman, A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 13–37. [Google Scholar]
- Lara-Diaz, V.; Castilla-Cortazar, I.; Martín-Estal, I.; García-Magariño, M.; Aguirre, G.; Puche, J.; de la Garza, R.; Morales, L.; Muñoz, U. IGF-1 modulates gene expression of proteins involved in inflammation, cytoskeleton, and liver architecture. J. Physiol. Biochem. 2017, 73, 245–258. [Google Scholar] [CrossRef]
- Lupu, F.; Terwilliger, J.D.; Lee, K.; Segre, G.V.; Efstratiadis, A. Roles of Growth Hormone and Insulin-like Growth Factor 1 in Mouse Postnatal Growth. Dev. Biol. 2001, 229, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Agrogiannis, G.D.; Sifakis, S.; Patsouris, E.S.; Konstantinidou, A.E. Insulin-like growth factors in embryonic and fetal growth and skeletal development (Review). Mol. Med. Rep. 2014, 10, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, B.L.; Vasishta, R.K.; Radotra, B.D. Myopathology: Common Terminologies Illustrated. In Myopathology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 73–97. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence | Temperature (°C) |
|---|---|---|
| IGF-1-Forward | GCTGGTGGATGCTCTTCAGT | 60 |
| IGF-1-Reverse | ACCCTGTGGGCTTGTTGAAA | 62 |
| POLG1-Forward | GAGAAGGCCCAGCAGATGTA | 60 |
| POLG1-Reverse | ATCCGACAGCCGATACCA | 60 |
| PPIA-Forward | TTTCATCTGCACTGCCAAGA | 60 |
| PPIA-Reverse | TTGCCAAACACCACATGCT | 61 |
| RPLP0-Forward | TGCATCAGTACCCCATTCTATCA | 61 |
| RPLP0-Reverse | AAGGTGTAATCCGTCTCCACAGA | 62 |
| Patients | |||||
|---|---|---|---|---|---|
| Clinical Signs | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 |
| Ethnicity | Tunisia: Bizerte | Tunisia: Bizerte | Tunisia: Bizerte | Tunisia: Kebeli | Tunisia: Tataouin |
| Gender (Male/Female) | Female | Female | Female | Female | Female |
| Age (Years) | 10 | 15 | 23 | 9 | 9 |
| Number of Cases Within the Same Family | 1 | 2 | 1 | 1 | 1 |
| Consanguinity | Endogamy | Endogamy | Consanguineous | Consanguineous | Consanguineous |
| Molecular Diagnosis | HTZ CHRNG gene (NM_005199.5) exon 5 c.351-1G > A/ rs: 761413806 | HMZ TPM2 gene (NM_003289.4); exon 6 c.628C > T; p.Q210*/rs199476154 | HMZ TPM2 gene (NM_003289.4); exon 6 c.628C > T; p.Q210*/rs199476154 | HMZ CHRNG gene (NM_005199.5) exon 7 c.753_754del; p.Val253Alafs*44/rs767503038 | HMZ CHRNG gene (NM_005199.5) exon 7 c.753_754del; p.Val253Alafs*44/rs767503038 |
| History of Spontaneous Abortion/LMPS | − | + | + | + | + |
| Short Stature | + | + | + | − | + |
| Height (m) | 1.35 | 1.40 | 1.35 | 1.30 | 1.20 |
| Weight (kg) | 50 | 46 | 45 | 26 | 12 |
| Reduced Fetal Movement | NI | NI | + | + | NI |
| Facial Dysmorphism | Ptosis, small mouth, low nose bridge | Ptosis, small mouth, low nose bridge | Ptosis, small mouth, low nose bridge | Ptosis, small mouth, low nose bridge | Ptosis, small mouth |
| Short Neck | + | + | + | + | + |
| Low-Set Ears | + | + | + | + | + |
| Motor Development | Delayed | Delayed | Delayed | Delayed | Delayed |
| Walking Impairment | − | +(with assistance) | +(wheelchair) | − | − |
| Arthrogryposis | + | + | + | + | + |
| Multiple Pterygium | + | + | + | + | + |
| Neck Pterygium | + | + | + | + | + |
| Axilla Pterygium | + | + | + | + | + |
| Elbows Pterygium | + | + | + | + | + |
| Knees Pterygium | + | + | + | + | + |
| Camptodactyly | + | + | + | + | + |
| Bilateral Vertical Talus | + | + | + | + | + |
| Respiratory Involvement | − | − | + | − | − |
| Scoliosis | Lumbar scoliosis | Severe dorsal scoliosis | Lordoscoliosis | − | Lordoscoliosis |
| Scoliosis: Cob Angle before Treatment | 60° | 80° | −50° | − | 25° |
| Orthopedic treatment/Physiotherapy | Bracing physiotherapy | Bracing Physiotherapy and knee splinting | Physiotherapy and knee splinting Halo-gravity traction | Orthopedic shoes | |
| Surgical Treatment For: | + | + | + | + | + |
| Flexion Contracture of the Knees | Ilizarov external fixator Distal femur osteotomy | Bilateral soft-tissue release Distal femur osteotomy | Bilateral soft-tissue release | Distal femur osteotomy | Distal femur osteotomy |
| Bilateral Vertical Talus | + | + | + | + | |
| Hip Dislocation | Soft-tissue release | − | − | + pelvic osteotomy | |
| Surgical Intervention for the Correction of the Scoliosis | Dual traditional growing rods and pelvic fixation Spinal arthrodesis | Single traditional growing rods | Anterior and posterior arthrodesis | − | |
| Cob Angle of the Scoliosis after Surgical | 40° | 40° | −20° | − | − |
| Conclusion of the EMG | Lower acceptable range for the nerve conductance and velocity | Lower acceptable range for the nerve conductance and velocity | NI | Lower acceptable range for the nerve conductance and velocity | NI |
| Other | - Pelvic obliquity - Pterygium in the groin and inguinal region | - Reduced muscle bulk and fatigability in lower limbs | - Impaired sitting ability - Reduced muscle bulk and fatigability in lower limbs | - Bilateral hip dislocation - Pterygium in the groin and inguinal region | − |
| Prediction Site | Score | Significance |
|---|---|---|
| Mutation Taster | 1 | Pathogenic |
| Human Splicing Finder | 55.45 | Alteration of the site |
| Maximum entropy model | −2.58 | Not predicted as a splice site |
| Markov Model | −2.61 | Not predicted as a splice site |
| Weight Matrix Model | −3.83 | Not predicted as a splice site |
| VarSEAK | −3.27% (class5 splicing affect) | exon skipping, loss of function |
| Variation in the Different Gene Pairs | Classification Score | Support Score | Predicted Class | Confidence Zone |
|---|---|---|---|---|
| KLHL40 CACNA1S | 0.8671 | 100.00 | Disease causing | 99% confidence |
| KLHL30 CACNA1S | 0.7400 | 99.20 | Disease causing | >95% confidence |
| TPM2 CACNA1S | 0.5500 | 55.00 | Disease causing | 90% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najjar, D.; Chikhaoui, A.; Zarrouk, S.; Azouz, S.; Kamoun, W.; Nassib, N.; Bouchoucha, S.; Yacoub-Youssef, H. Combining Gene Mutation with Expression of Candidate Genes to Improve Diagnosis of Escobar Syndrome. Genes 2022, 13, 1748. https://doi.org/10.3390/genes13101748
Najjar D, Chikhaoui A, Zarrouk S, Azouz S, Kamoun W, Nassib N, Bouchoucha S, Yacoub-Youssef H. Combining Gene Mutation with Expression of Candidate Genes to Improve Diagnosis of Escobar Syndrome. Genes. 2022; 13(10):1748. https://doi.org/10.3390/genes13101748
Chicago/Turabian StyleNajjar, Dorra, Asma Chikhaoui, Sinda Zarrouk, Saifeddine Azouz, Wafa Kamoun, Nabil Nassib, Sami Bouchoucha, and Houda Yacoub-Youssef. 2022. "Combining Gene Mutation with Expression of Candidate Genes to Improve Diagnosis of Escobar Syndrome" Genes 13, no. 10: 1748. https://doi.org/10.3390/genes13101748