
Comparative and Phylogenetic Analyses of Complete Chloroplast Genomes of Scrophularia incisa Complex (Scrophulariaceae)
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
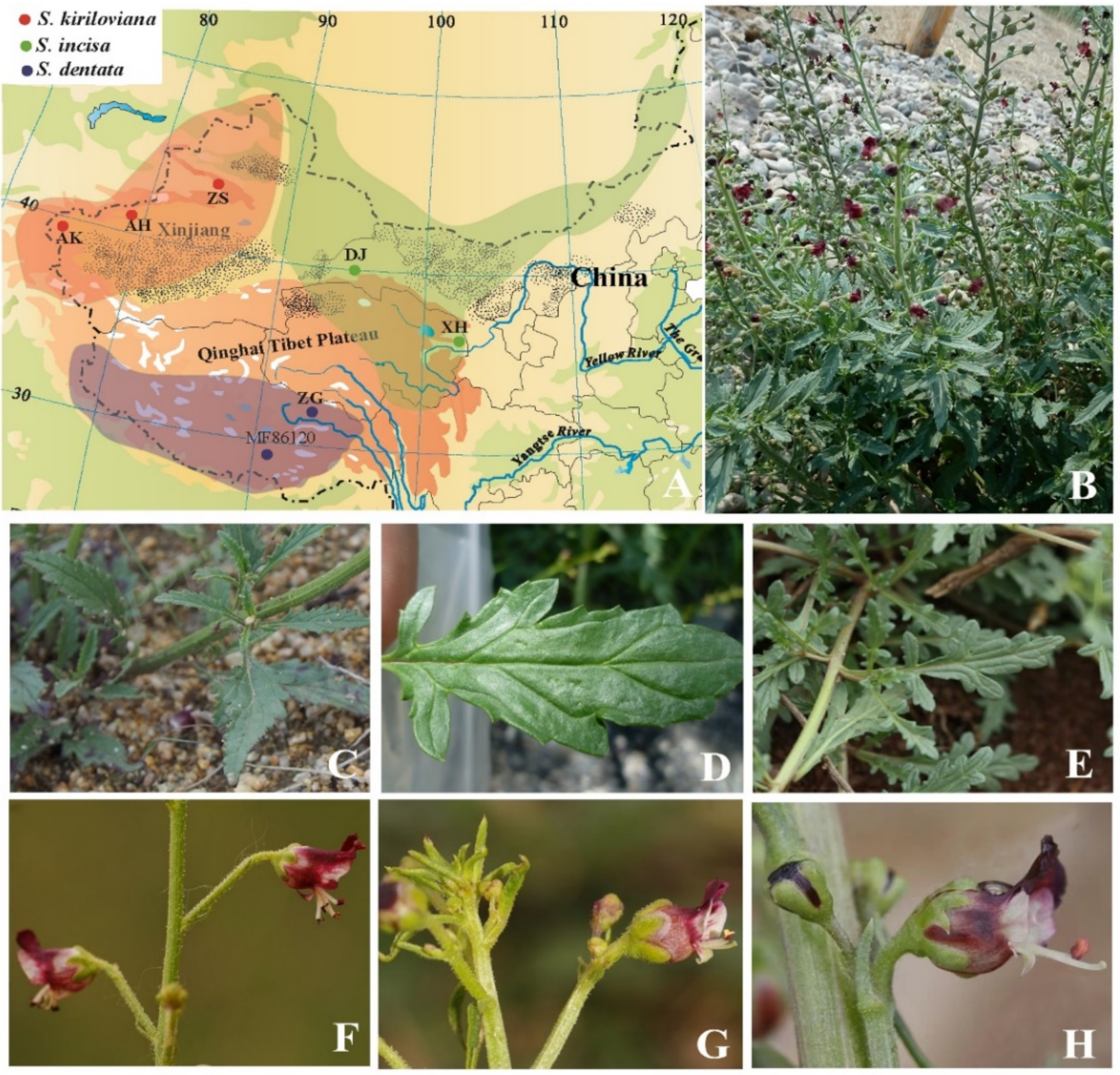
2.1. Plant Samples, Sequencing and Assembly
2.2. Chloroplast Genome Annotation, Comparison and Codon Usage Bias Analysis
2.3. Characterization of Repeat Sequences and Simple Sequence Repeats
2.4. Selective Pressure Analysis
2.5. Phylogenetic Analysis
3. Results
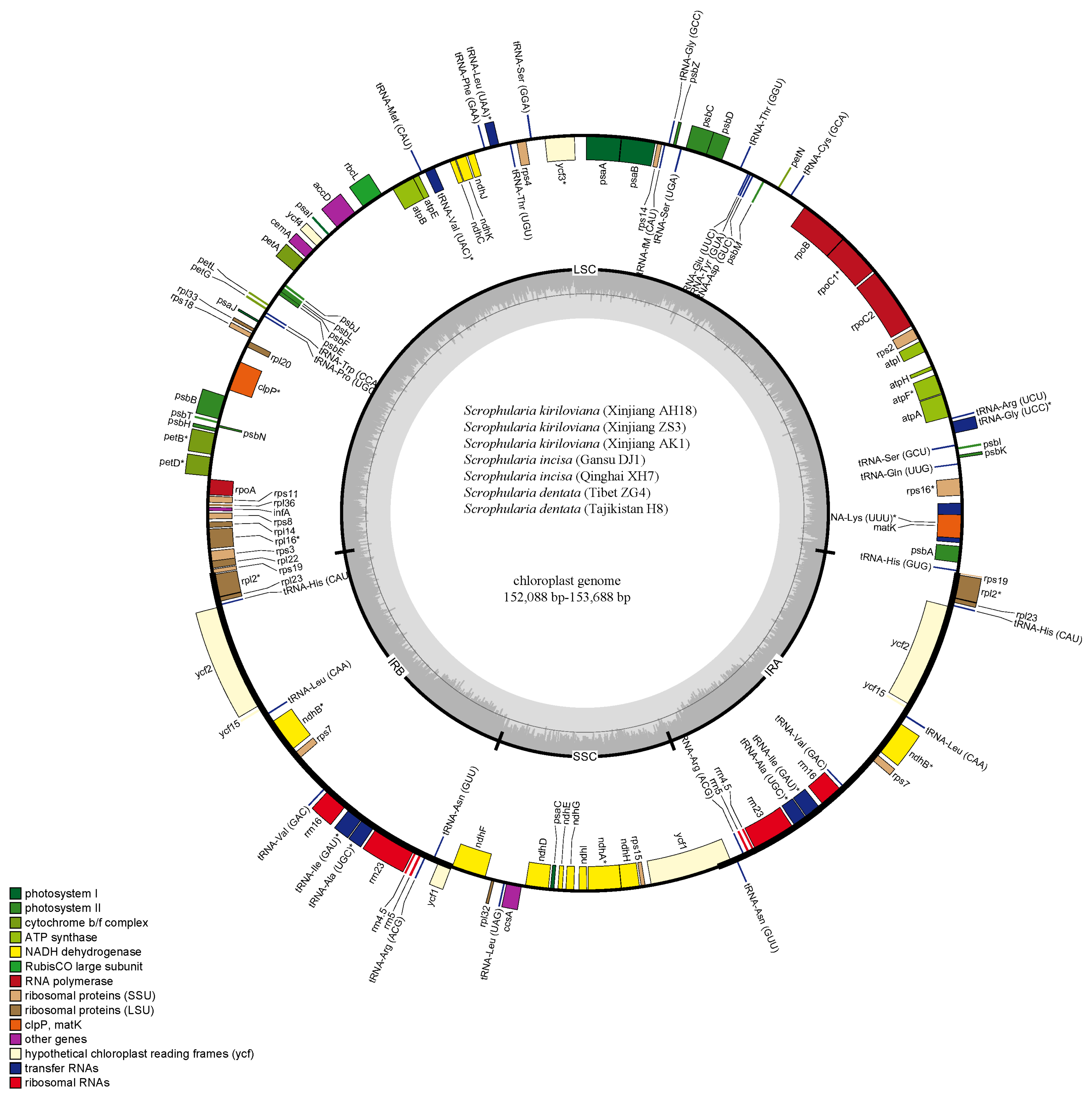
3.1. Genome Organization and Features
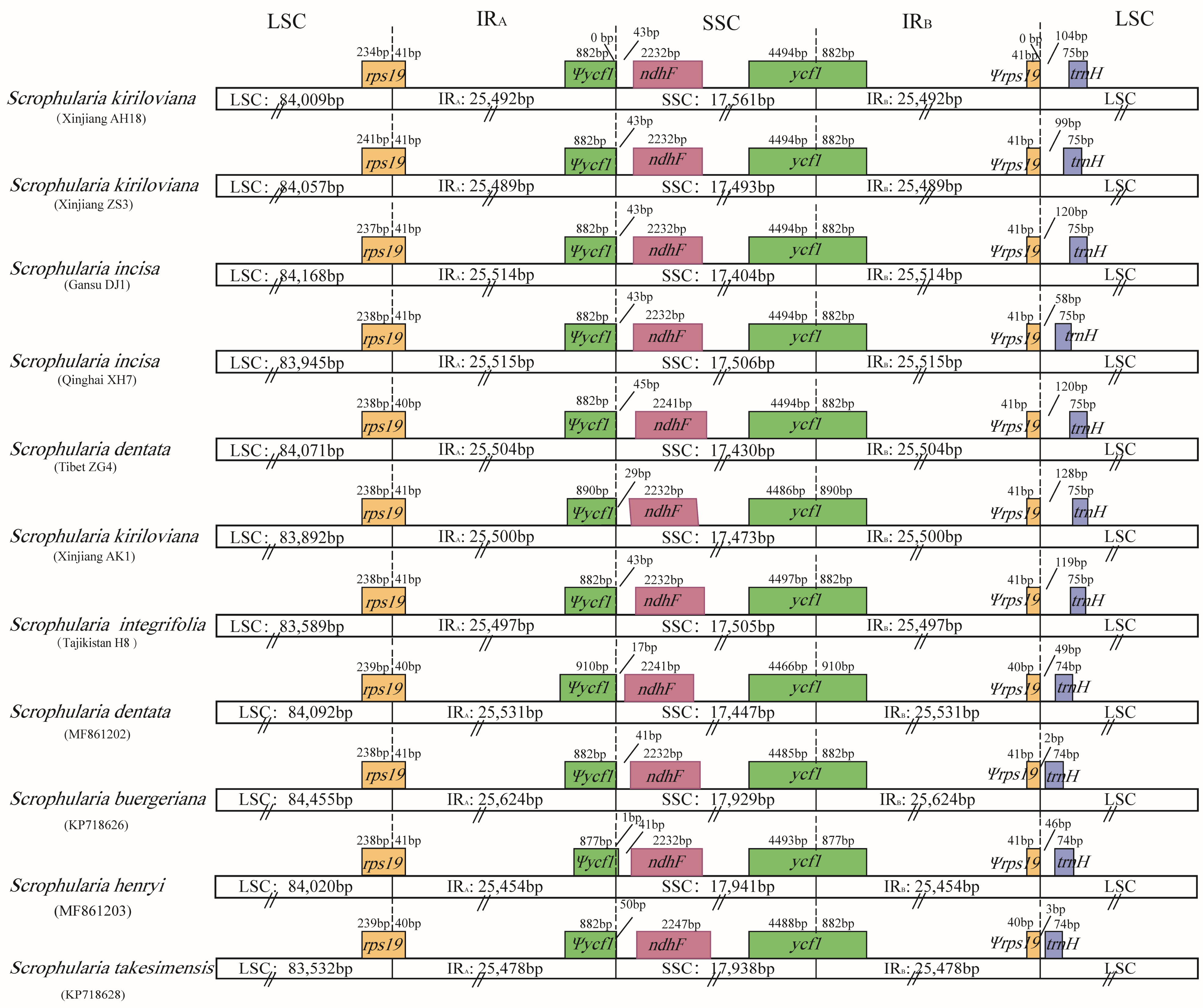
3.2. Contraction and Expansion of Inverted Repeats (IRs)
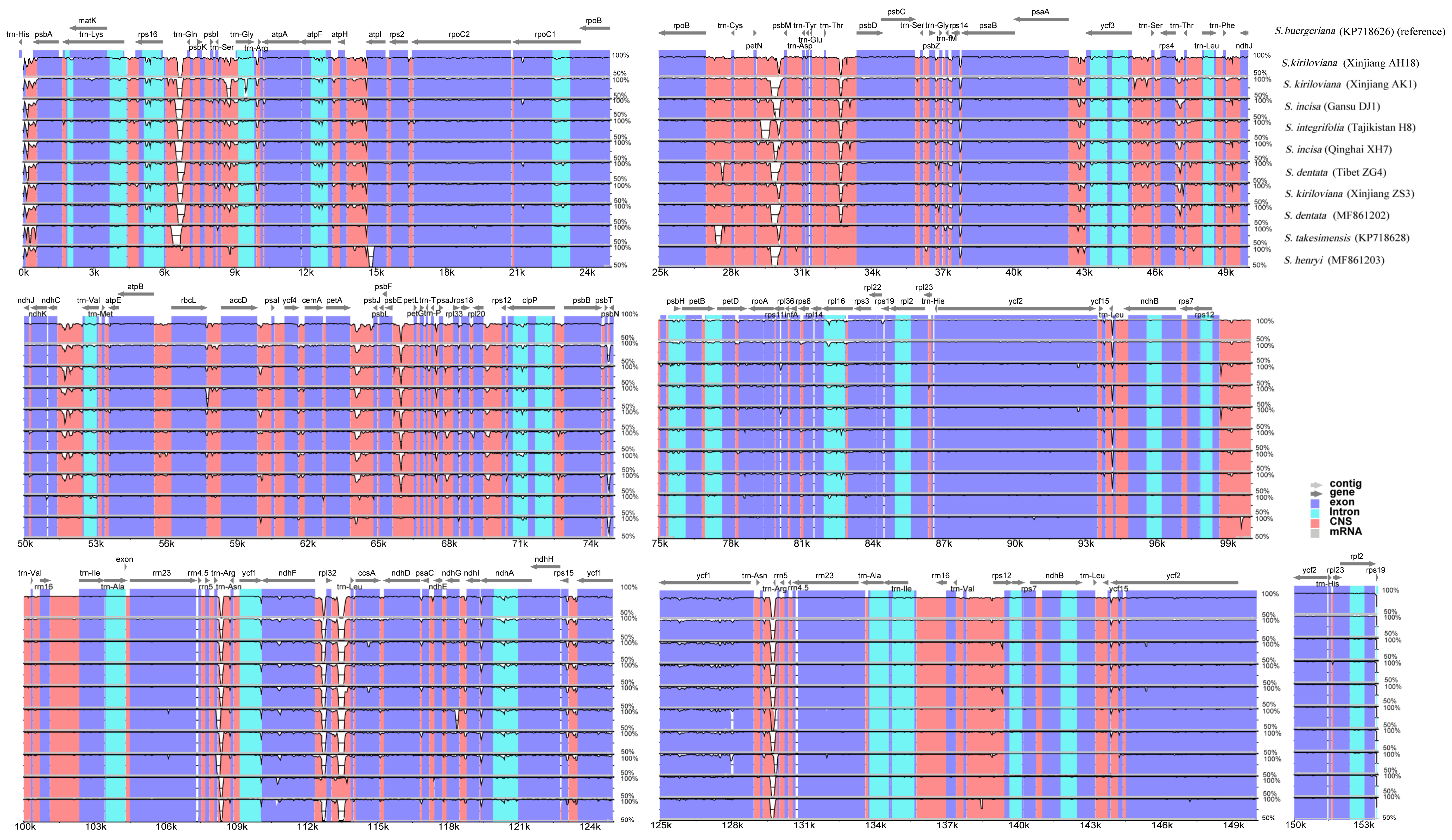
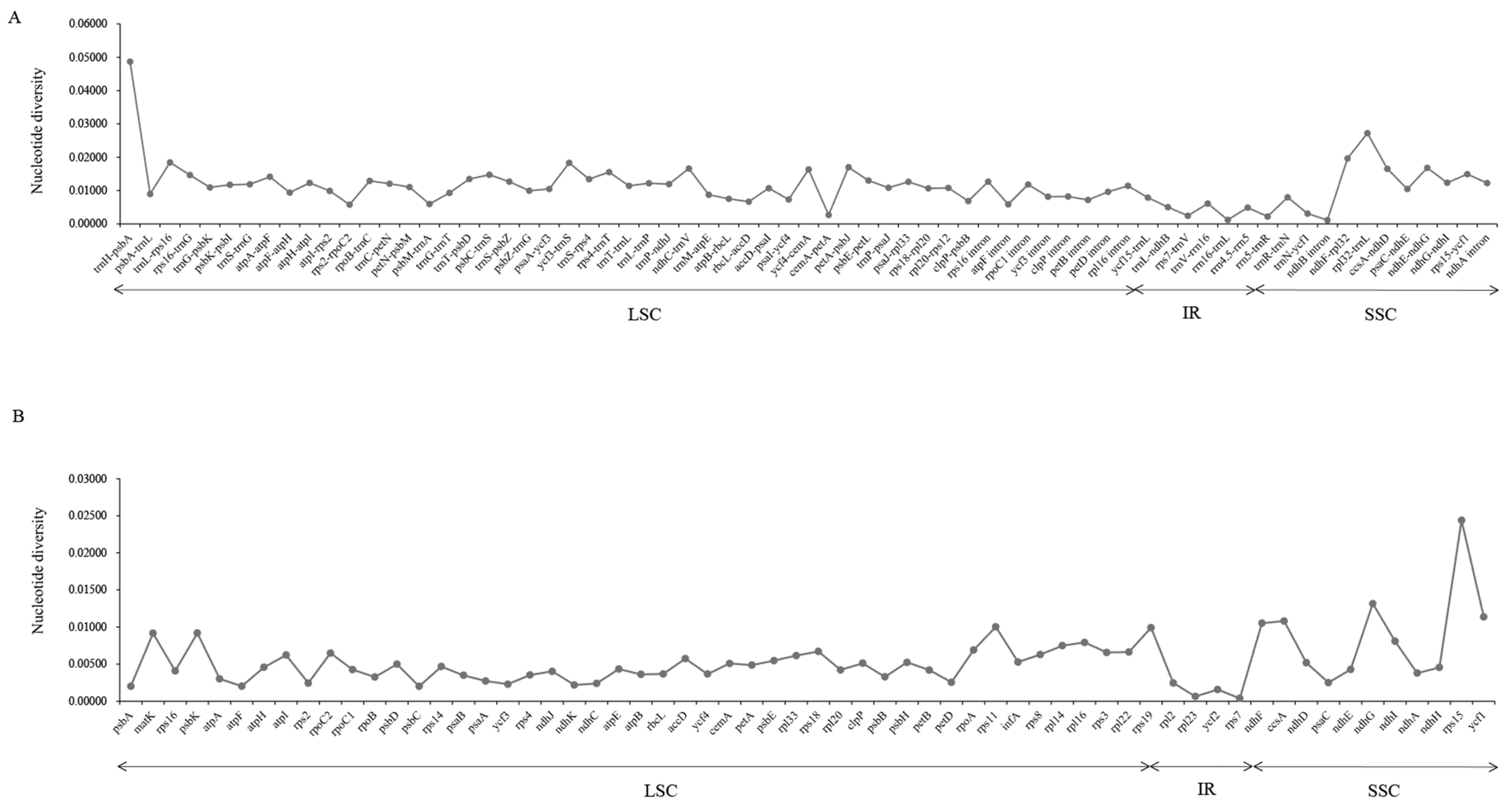
3.3. Genome Comparative Analysis and Molecular Marker Identification
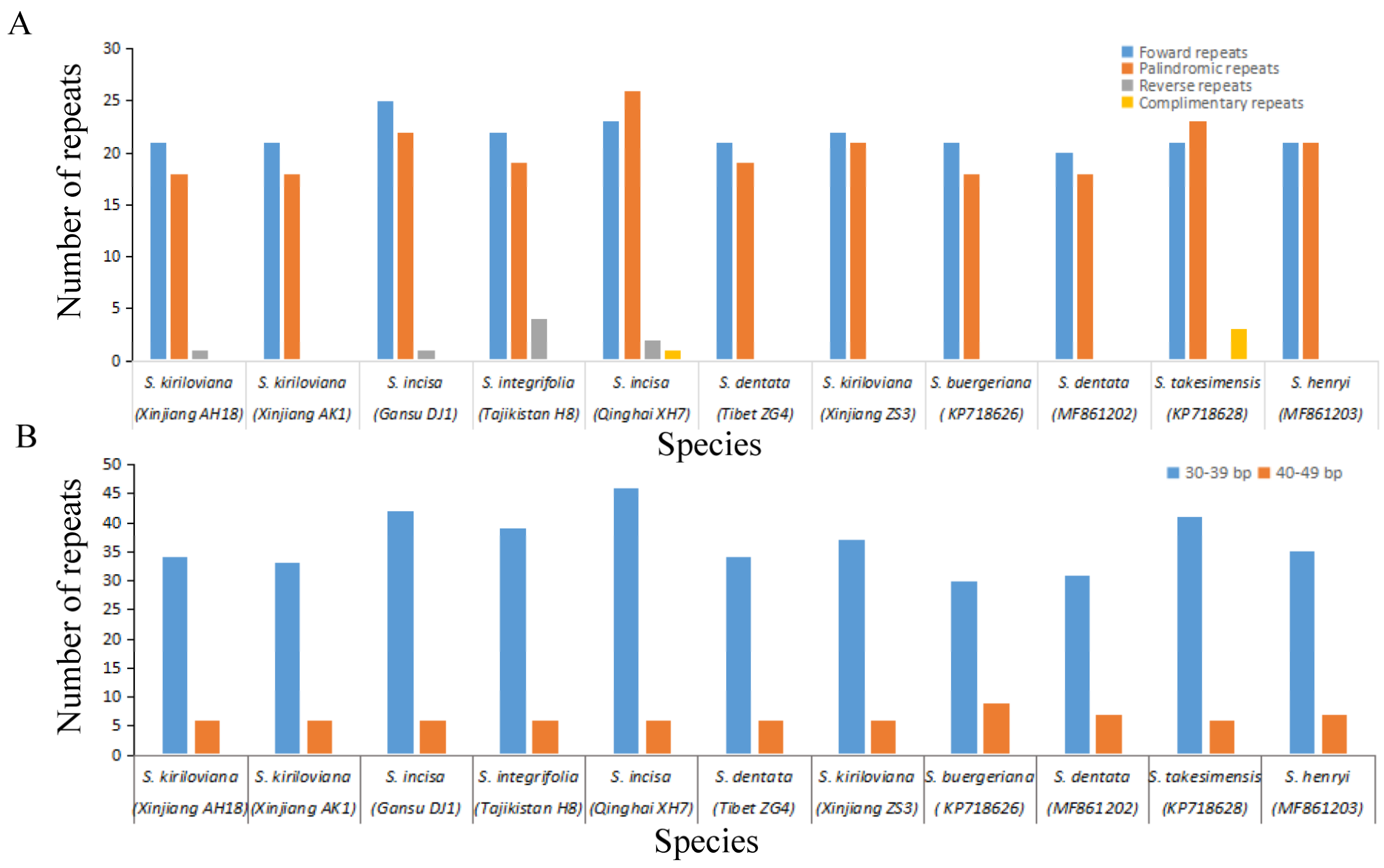
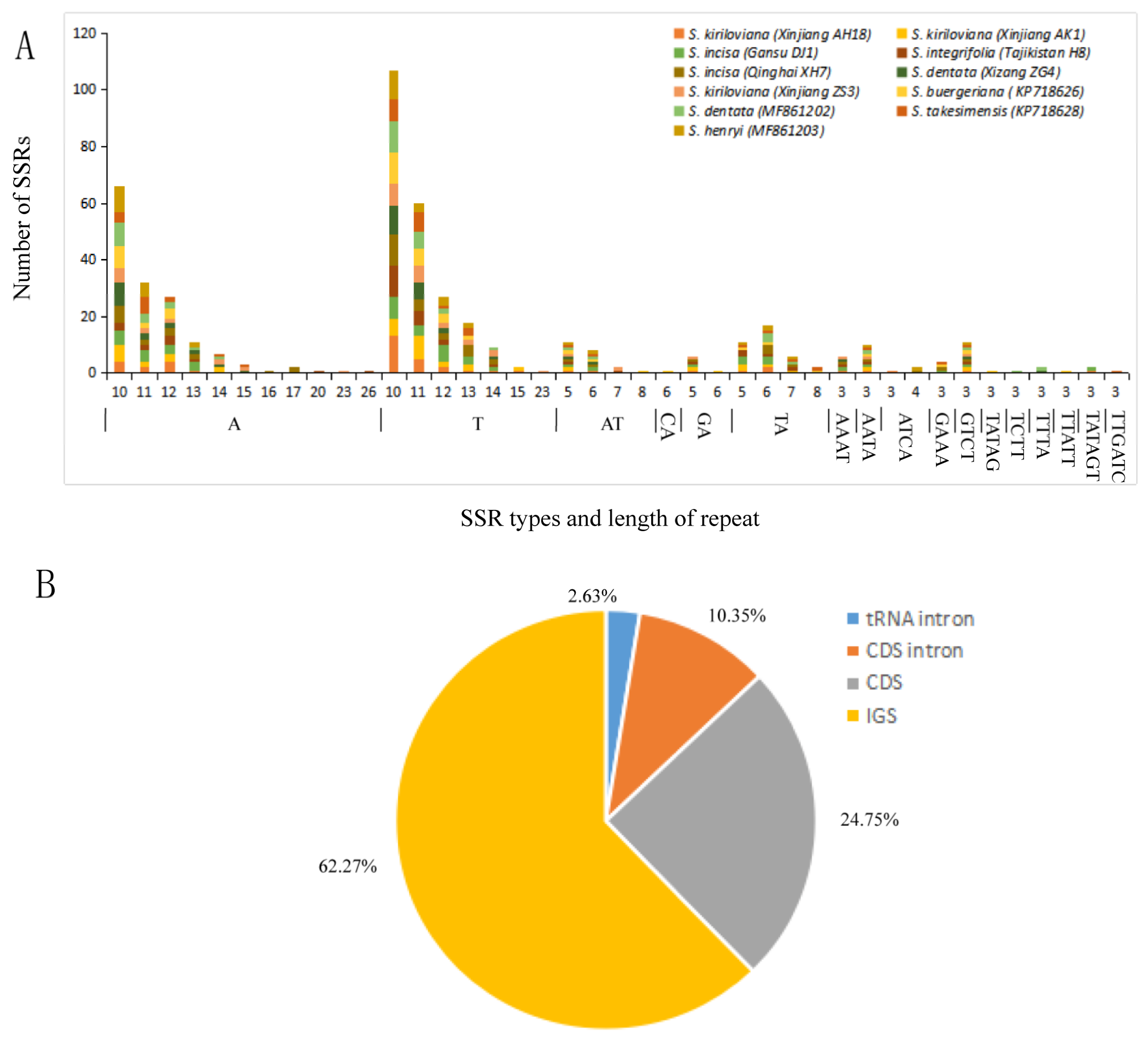
3.4. Characterization of Repeat Sequences and SSRs
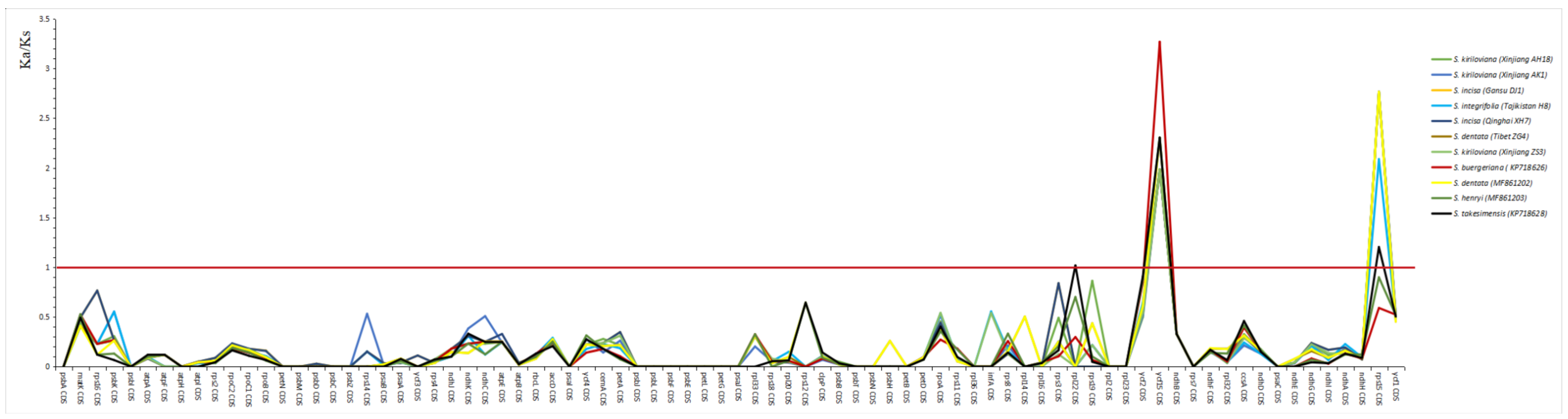
3.5. Selective Pressure Analysis
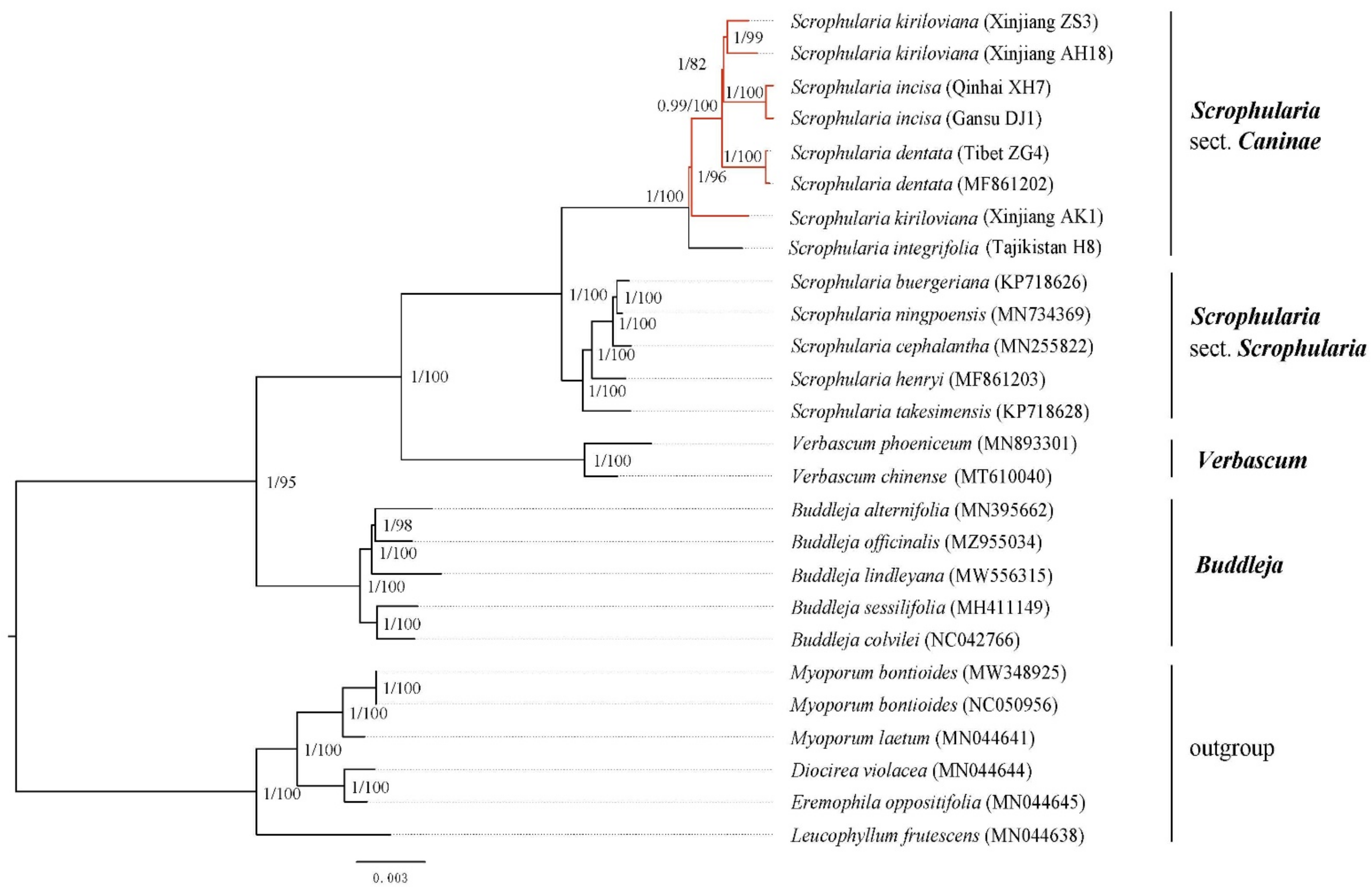
3.6. Phylogenetic Analyses
4. Discussion
4.1. Comparative Genomics
4.2. Phylogenetic Relationships within S. incisa Complex and Scrophulariaceae
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hong, D.-Y. The Distribution of Scrophulariaceae in the Holarctic with Special Reference to the Floristic Relationships Between Eastern Asia and Eastern North America. Ann. Mo. Bot. Gard. 1983, 70, 701. [Google Scholar] [CrossRef]
- Willis, J.C. A Dictionary of the Flowering Plants and Ferns. Q. Rev. Biol. 1974, 49, 436. [Google Scholar] [CrossRef]
- Mabberley, D.J. The Plant-Book: A Portable Dictionary of the Vascular Plants; Cambridge University Press: Cambridge, UK, 1997; p. 169. [Google Scholar]
- Fischer, E. Scrophulariaceae. In The Families and Genera of Vascular Plants; Kadereit, J.W., Ed.; Springer: Berlin/Heidelberg, Germany, 2004; Volume 7, pp. 333–432. [Google Scholar]
- Scheunert, A.; Heubl, G. Against all odds: Reconstructing the evolutionary history of Scrophularia (Scrophulariaceae) despite high levels of incongruence and reticulate evolution. Org. Divers. Evol. 2017, 17, 323–349. [Google Scholar] [CrossRef]
- Tsoong, P.C.; Tang, Y.C. Scrophularia. In Flora Reipublicae Popularis Sinicae, Tomus; Tsoong, P.C., Yang, H.B., Eds.; Science Press: Beijing, China, 1979; Volume 67, pp. 46–85. [Google Scholar]
- Hong, D.Y.; Yang, H.B.; Jin, C.L.; Holmgren, N.H. Scrophulariaceae. In Flora of China; Wu, Z.-Y., Ed.; Missouri Botanical Garden: St. Louis, MI, USA, 1998; Volume 18, pp. 1–212. [Google Scholar]
- Stiefelhagen, H. Systematische und Pflanzengeographische Studien zur Kenntnis der Gattung Scrophularia. Bot. Jahrbücher Syst. Pflanzengesch. Pflanzengeogr. 1910, 44, 406–496. [Google Scholar]
- Wang, R.H.; Xia, M.Q.; Tan, J.B.; Chen, C.; Jin, X.J.; Li, P.; Fu, C.X. A new species of Scrophularia (Scrophulariaceae) from Hubei, China. Phytotaxa 2018, 350, 1–14. [Google Scholar] [CrossRef]
- Scheunert, A.; Heubl, G. Phylogenetic relationships among New World Scrophularia L. (Scrophulariaceae): New insights inferred from DNA sequence data. Plant Syst. Evol. 2011, 291, 69–89. [Google Scholar] [CrossRef]
- Faride, A.; Mehrshid, R.; Fatemeh, D.; Fatemeh, A.; Fatemeh, A. Preliminary molecular phylogeny of Eurasian Scrophularia (Scrophulariaceae) based on DNA sequence data from trnS-trnG and ITS regions. G. Bot. Ital. 2011, 145, 857–865. [Google Scholar] [CrossRef]
- Navarro-Pérez, M.L.; López, L.; Fernández-Mazuecos, M.; Rodríguez-Riaño, T.; Ortega-Olivencia, A. The role of birds and insects in pollination shifts of Scrophularia (Scrophulariaceae). Mol. Phylogenet. Evol. 2013, 69, 239–254. [Google Scholar] [CrossRef]
- Scheunert, A.; Heubl, G. Diversification of Scrophularia (Scrophulariaceae) in the Western Mediterranean and Macaronesia–Phylogenetic relationships, reticulate evolution and biogeographic patterns. Mol. Phylogenet. Evol. 2014, 70, 296–313. [Google Scholar] [CrossRef]
- Chen, C.; Cai, M.Q.; Xu, B.; Jin, X.J.; Wang, R.H.; Li, P.; Zhao, Y.P.; Fu, C.X. Systematic position of Oreosolen (tribe Scrophularieae, Scrophulariaceae) based on nuclear and plastid sequences, J. Syst. Evol. 2017, 55, 446–452. [Google Scholar] [CrossRef]
- Wang, R.H.; Wang, D.D.; Li, P.; Qi, Z.C.; Fu, C.X. Scrophularia koraiensis, a new synonym to Scrophularia kakudensis (Lamiales: Scrophulariaceae). Phytotaxa 2015, 202, 228–230. [Google Scholar] [CrossRef]
- Wang, R.H.; Chen, C.; Ma, Q.; Li, P.; Fu, C.X. Development of microsatellite loci in Scrophularia incisa (Scrophulariaceae) and cross-amplification in congeneric species. Appl. Plant Sci. 2014, 2, 1300077. [Google Scholar] [CrossRef]
- Wang, R.H.; Yang, Z.P.; Zhang, Z.C.; Comes, H.P.; Qi, Z.C.; Li, P.; Fu, C.X. Plio-Pleistocene climatic change drives allopatric speciation and population divergence within Scrophularia incisa complex (Scrophulariaceae) of the desert and steppe subshrubs in Northwest China. Front. Plant Sci. 2022, 13, 985372. [Google Scholar] [CrossRef]
- Zhang, L.Q.; Yang, Z.; Jia, Q.; Dorje, G.; Zhao, Z.; Guo, F.J.; Li, Y.M. Two new phenylpropanoid glycosides with interesterification from Scrophularia dentata Royle ex Benth. J. Mol. Struct. 2013, 1049, 299–302. [Google Scholar] [CrossRef]
- Zhang, L.Q.; Zhu, T.T.; Qian, F.; Xu, J.W.; Dorje, G.; Zhao, Z.L.; Guo, F.J.; Li, Y.M. Iridoid glycosides isolated from Scrophularia dentata Royle ex Benth. and their anti-inflammatory activity. Fitoterapia 2014, 98, 84–90. [Google Scholar] [CrossRef]
- Ullah, F.; Gao, Y.; Sari, I.; Jiao, R.F.; Saqib, S.; Gao, X.F. Macro-Morphological and Ecological Variation in Rosa sericea Complex. Agronomy 2022, 12, 1078. [Google Scholar] [CrossRef]
- Wang, R.H. Phylogeny and Biogeography of Scrophularia and Phylogeography of S. incisa Complex; Zhejiang University: Hangzhou, China, 2015. [Google Scholar] [CrossRef]
- Liu, L.X.; Li, R.; Worth, J.R.P.; Li, X.; Li, P.; Cameron, K.M.; Fu, C.X. The Complete Chloroplast Genome of Chinese Bayberry (Morella rubra, Myricaceae): Implications for Understanding the Evolution of Fagales. Front. Plant Sci. 2017, 8, 968. [Google Scholar] [CrossRef]
- Ran, H.; Liu, Y.Y.; Wu, C.; Cao, Y.N. Phylogenetic and comparative analyses of complete chloroplast genomes of Chinese Viburnum and Sambucus (Adoxaceae). Plants 2020, 9, 1143. [Google Scholar] [CrossRef]
- Burke, S.V.; Grennan, C.P.; Duvall, M.R. Plastome sequences of two New World bamboos—Arundinaria gigantea and Cryptochloa strictiflora (Poaceae)—Extend phylogenomic understanding of Bambusoideae. Am. J. Bot. 2012, 99, 1951–1961. [Google Scholar] [CrossRef]
- Sokolowska, J.; Fuchs, H.; Celinski, K. New insight into taxonomy of European Mountain Pines, Pinus mugo complex, based on complete chloroplast genomes sequencing. Plants 2021, 10, 1331. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Guo, Y.; Chen, S.; Wang, R. Complete chloroplast genomes of Leptodermis scabrida complex: Comparative genomic analyses and phylogenetic relationships. Gene 2021, 791, 145715. [Google Scholar] [CrossRef]
- Lu, R.S.; Li, P.; Qiu, Y.X. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: Comparative genomic and phylogenetic analyses. Front. Plant Sci. 2017, 7, 2054. [Google Scholar] [CrossRef]
- Li, P.; Lu, R.S.; Xu, W.Q.; Ohi-Toma, T.; Cai, M.Q.; Qiu, Y.X.; Cameron, K.M.; Fu, C.X. Comparative Genomics and Phylogenomics of East Asian Tulips (Amana, Liliaceae). Front. Plant Sci. 2017, 8, 451. [Google Scholar] [CrossRef]
- Ye, W.Q.; Yap, Z.Y.; Li, P.; Comes, H.P.; Qiu, Y.X. Plastome organization, genome-based phylogeny and evolution of plastid genes in Podophylloideae (Berberidaceae). Mol. Phylogenet. Evol. 2018, 127, 978–987. [Google Scholar] [CrossRef]
- Cronn, R.; Liston, A.; Parks, M.; Gernandt, D.S.; Shen, R.; Mockler, T. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 2008, 36, e122. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes witDOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef]
- Feng, J.Y.; Wu, Y.Z.; Wang, R.R.; Xiao, X.F.; Wang, R.H.; Qi, Z.C.; Yan, X.L. The complete chloroplast genome of balsam aster (Aster ageratoides Turcz. var. scaberulus (Miq.) Ling., Asteraceae). Mitochondrial DNA Part B 2021, 6, 2464–2465. [Google Scholar] [CrossRef]
- Zhang, C.J.; Xia, P.G.; Wu, R.; Mans, D. The complete chloroplast genome of Scutellaria meehanioides (Lamiaceae) from Shaanxi Province, China. Mitochondrial DNA Part B 2021, 6, 1685–1686. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, M.M.; Zhang, X.M.; Chen, S.N.; Liang, Z.S. The complete chloroplast genome sequence of traditional Chinese medicine Uncaria macrophylla (Rubiaceae). Mitochondrial DNA B Resour. 2022, 7, 694–695. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, 686–689. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, 575–581. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Liu, X.S. Patterns and influencing factor of synonymous codon usage in porcine circovirus. Virol. J. 2012, 9, 68. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Kurtz, S.; Schleiermacher, C. REPuter: Fast computation of maximal repeats in complete genomes. Bioinformatics 1999, 15, 426–427. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Ni, L.H.; Zhao, Z.L.; Dorje, G.; Ma, M. The Complete Chloroplast Genome of Ye-Xing-Ba (Scrophularia dentata; Scrophulariaceae), an Alpine Tibetan Herb. PLoS ONE 2016, 11, e0158488. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. Chloroplast evolution: Secondary symbiogenesis and multiple losses. Curr. Biol. 2002, 12, R62–R64. [Google Scholar] [CrossRef]
- Kim, P.M.; Korbel, J.O.; Gerstein, M.B. Positive selection at the protein network periphery: Evaluation in terms of structural constraints and cellular context. Proc. Natl. Acad. Sci. USA 2007, 104, 20274–20279. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Ponce, D.; Aguade, M.; Rozas, J. Network-level molecular evolutionary analysis of the insulin/TOR signal transduction pathway across 12 Drosophila genomes. Genome Res. 2008, 19, 234–242. [Google Scholar] [CrossRef]
- Kosiol, C.; Vinař, T.; Fonseca, R.R.; Hubisz, M.J.; Bustamante, C.D.; Nielsen, R.; Siepel, A. Patterns of positive selection in six Mammalian genomes. PLoS Genet. 2008, 4, e1000144. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef]
- Xu, W.Q.; Losh, J.; Chen, C.; Li, P.; Wang, R.H.; Zhao, Y.P.; Qiu, Y.X.; Fu, C.X. Comparative genomics of figworts (Scrophularia, Scrophulariaceae), with implications for the evolution of Scrophularia and Lamiales. J. Syst. Evol. 2019, 57, 55–65. [Google Scholar] [CrossRef]
- Bi, Y.Q.; Deng, P.; Liu, L.X. The complete chloroplast genome sequence of purple mullein (Verbascum phoeniceum L.). Mitochondrial DNA Part B 2020, 5, 819–820. [Google Scholar] [CrossRef]
- Fowler, R.M.; Mclay, T.G.B.; Schuster, T.M.; Buirchell, B.J.; Murphy, D.J.; Bayly, M.J. Plastid phylogenomic analysis of tribe Myoporeae (Scrophulariaceae). Plant Syst. Evol. 2020, 306, 52. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Alexandros, S. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Zang, M.; Li, M.; Fang, Y. Complete chloroplast genome sequence and phylogenetic analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Qi, X.; Chen, J.; Sun, L.; Zhong, Y.; Fang, J.; Hu, C. The complete chloroplast genome sequence of Actinidia arguta using the PacBio RS II platform. PLoS ONE 2018, 13, e0197393. [Google Scholar] [CrossRef]
- Timme, R.E.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot. 2007, 94, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.B.; Yang, S.X.; Li, H.T.; Yang, J.; Li, D.Z. Comparative Chloroplast Genomes of Camellia Species. PLoS ONE 2013, 8, e73053. [Google Scholar] [CrossRef]
- Song, Y.; Yao, X.; Tan, Y.H.; Gan, Y.; Corlett, R.T. Complete chloroplast genome sequence of the avocado: Gene organization, comparative analysis, and phylogenetic relationships with other Lauraceae. Can. J. For. Res. 2016, 46, 1293–1301. [Google Scholar] [CrossRef]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.; Wang, L.; Biradar, S.S.; Tan, X.; Wan, F.; Weining, S. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef]
- Sithichoke, T.; Pichahpuk, U.; Duangjai, S.; Juntima, C.; Thippawan, Y.; Nukoon, J.; Somvong, T. Characterization of the complete chloroplast genome of Hevea brasiliensis reveals genome rearrangement, RNA editing sites and phylogenetic relationships. Gene 2011, 475, 104–112. [Google Scholar] [CrossRef]
- Yi, D.K.; Kim, K.J. The two complete plastomes from Scrophularia (Scrophulariaceae): Scrophularia buergeriana and S. takesimensis. Mitochondrial DNA Part B 2016, 1, 710–712. [Google Scholar] [CrossRef]
- Nazareno, A.G.; Carlsen, M.; Lohmann, L.G. Complete Chloroplast Genome of Tanaecium tetragonolobum: The First Bignoniaceae Plastome. PLoS ONE 2015, 10, e0129930. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, L.; Liu, A.; Chen, J.J.; Wu, L.; Hu, W.M.; Zhang, W.; Kim, K.; Lee, S.C.; Yang, T.J.; et al. The Complete Chloroplast Genome Sequences of Five Epi medium Species: Lights into Phylogenetic and Taxonomic Analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Bodin, S.S.; Kim, J.S.; Kim, J.H. Complete Chloroplast Genome of Chionographis japonica (Willd.) Maxim. (Melanthiaceae): Comparative Genomics and Evaluation of Universal Primers for Liliales. Plant Mol. Biol. Rep. 2013, 31, 1407–1421. [Google Scholar] [CrossRef]
- Zhao, Y.; Yin, J.; Guo, H.; Zhang, Y.; Xiao, W.; Sun, C.; Wu, J.; Qu, X.; Yu, J.; Wang, X. The complete chloroplast genome provides insight into the evolution and polymorphism of Panax ginseng. Front. Plant Sci. 2015, 5, 696. [Google Scholar] [CrossRef]
- Mao, K.S.; Wang, Y.; Liu, J.Q. Evolutionary origin of species diversity on the Qinghai-Tibet Plateau. J. Syst. Evol. 2021, 59, 1142–1158. [Google Scholar] [CrossRef]
- Spicer, R.A. Tibet, the Himalaya, Asian monsoons and biodiversity–in what ways are they related? Plant Divers. 2017, 39, 5–16. [Google Scholar] [CrossRef]
- Miao, Y.F.; Herrmann, M.; Wu, F.L.; Yan, X.L.; Yang, S.L. What controlled Mid-Late Miocene long-term aridification in Central Asia—Global cooling or Tibetan Plateau uplift: A review. Earth Sci. Rev. 2012, 112, 155–172. [Google Scholar] [CrossRef]
- Xu, L.; Cao, M.T.; Wang, Q.C.; Xu, J.H.; Liu, C.L.; Ullah, N.; Li, J.J.; Hou, Z.N.; Liang, Z.S.; Zhou, W.J.; et al. Insights into the plateau adaptation of Salvia castanea by comparative genomic and WGCNA analyses. J. Adv. Res. 2022. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Jansen, R.K. Chloroplast genomes of plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; Henry, R.J., Ed.; CABI Publishing: Wallingford, UK, 2005; pp. 45–68. [Google Scholar]
- Szczecińska, M.; Sawicki, J. Genomic Resources of Three Pulsatilla Species Reveal Evolutionary Hotspots, Species-Specific Sites and Variable Plastid Structure in the Family Ranunculaceae. Int. J. Mol. Sci. 2015, 16, 22258–22279. [Google Scholar] [CrossRef]
- Ma, J.; Yang, B.; Zhu, W.; Sun, L.; Tian, J.; Wang, X. The complete chloroplast genome sequence of Mahonia bealei (Berberidaceae) reveals a significant expansion of the inverted repeat and phylogenetic relationship with other angiosperms. Gene 2013, 528, 120–131. [Google Scholar] [CrossRef]
- Davis, J.I.; Soreng, R.J. Migration of endpoints of two genes relative to boundaries between regions of the plastid genome in the grass family (Poaceae). Am. J. Bot. 2010, 97, 874–892. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.R.; Dastidar, S.G.; Cai, Z.; Penaflor, C.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae). Mol. Phylogenet. Evol. 2007, 45, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, H. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat/large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2010, 46, 85–94. [Google Scholar] [CrossRef]
- Abbott, R.J.; Smith, L.C.; Milne, R.I.; Crawford, R.M.M.; Wolff, K.; Balfour, J. Molecular analysis of plant migration and refugia in the Arctic. Science 2000, 289, 1343–1346. [Google Scholar] [CrossRef]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: London, UK, 2000. [Google Scholar]
- Zaman, W.; Ye, J.; Ahmad, M.; Saqib, S. Phylogenetic exploration of traditional Chinese medicinal plants: A case study on Lamiaceae. Pak. J. Bot. 2022, 54, 1033–1040. [Google Scholar] [CrossRef]
- Oxelman, B.; Kornhall, P.; Olmstead, R.G.; Bremer, B. Further disintegration of Scrophulariaceae. Taxon 2005, 54, 411–425. [Google Scholar] [CrossRef]
- Tank, D.C.; Beardsley, P.M.; Kelchner, S.A.; Olmstead, R.G. Review of the systematics of Scrophulariaceae s. l. and their current disposition. Austral. Syst. Bot. 2006, 19, 289–307. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Guan, K.; Zhou, Z.; Olmstead, R.; Cronk, Q. Molecular phylogeny of Incarvillea (Bignoniaceae) based on ITS and trnL–F sequences. Am. J. Bot. 2005, 92, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, Y.F.; Liu, S.B.; Huang, H.W. Molecular phylogeny of Myricaria (Tamaricaceae): Implications for taxonomy and conservation in China. Bot. Aquat. 2009, 50, 343–352. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, J.Q.; Nie, Z.L.; Zhong, Y.; Sun, H. Evolutionary diversifications of plants on the Qinghai-Tibetan Plateau. Front. Genet. 2014, 5, 4. [Google Scholar] [CrossRef]
- Yue, J.P.; Sun, H.; David, B.A.; Li, J.H.; Al-Shehbaz, I.A.; Ree, R. Molecular phylogeny of Solms-laubachia (Brassicaceae) s.l., based on multiple nuclear and plastid DNA sequences, and its biogeographic implications. J. Syst. Evol. 2009, 47, 402–415. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | S. integrifolia | S. kiriloviana | S. kiriloviana | S. kiriloviana | S. incisa | S. incisa | S. dentata | S. dentata | S. buergeriana | S. takesimensis | S. henryi |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (Tajikistan H8) | (Xinjiang AH18) | (Xinjiang AK1) | (Xinjiang ZS3) | (Gansu DJ1) | (Qinghai XH7) | (Tibet ZG4) | |||||
| Total cpDNA size (bp) | 152,088 | 152,554 | 152,365 | 152,528 | 152,600 | 152,481 | 152,509 | 152,600 | 153,631 | 152,425 | 152,868 |
| Latitude (°N) | 36.617 | 40.746 | 38.762 | 43.197 | 39.633 | 35.662 | 29.490 | 30.653 | —— | —— | 31.470 |
| Longitude (°E) | 68.564 | 77.826 | 75.193 | 81.201 | 94.340 | 102.644 | 97.470 | 97.568 | —— | —— | 110.395 |
| LSC length | 83,589 | 84,009 | 83,892 | 84,057 | 84,168 | 83,945 | 84,071 | 84,091 | 84,454 | 83,531 | 84,020 |
| SSC length | 17,505 | 17,561 | 17,473 | 17,493 | 17,404 | 17,506 | 17,430 | 17,447 | 17,929 | 17,938 | 17,940 |
| IR length | 25,497 | 25,492 | 25,500 | 25,489 | 25,514 | 25,515 | 25,504 | 25,531 | 25,624 | 25,478 | 25,454 |
| Total GC content (%) | 38 | 38 | 38 | 38 | 37.9 | 38 | 38 | 38 | 38 | 38.1 | 38 |
| LSC | 36.1 | 36.1 | 36.1 | 36.1 | 36 | 36.1 | 36.1 | 36 | 43.2 | 36.2 | 36.1 |
| SSC | 32.1 | 32.1 | 32.1 | 32.2 | 32.1 | 32.1 | 32.2 | 32.2 | 32.2 | 32.2 | 32.2 |
| IR | 43.1 | 43.1 | 43.1 | 43.1 | 43.1 | 43.1 | 43.1 | 43.1 | 43.2 | 43.2 | 43.2 |
| Total number of genes | 132 | 132 | 132 | 132 | 132 | 132 | 132 | 132 | 132 | 132 | 132 |
| Protein-coding genes | 80 | 80 | 80 | 80 | 80 | 80 | 80 | 80 | 80 | 80 | 80 |
| rRNA genes | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| tRNA genes | 31 | 31 | 31 | 31 | 31 | 31 | 31 | 31 | 31 | 31 | 31 |
| Duplicated genes | 17 | 17 | 17 | 17 | 17 | 17 | 17 | 17 | 17 | 17 | 17 |
| GenBank Acc. No. | OP018678 | OP018676 | OP036427 | OP036428 | OP036429 | OP018675 | OP018677 | MF861202 | KP718626 | KP718628 | MF861203 |
| Groups of Gene | Name of Gene |
|---|---|
| Ribosomal RNAs | rrn16(×2), rrn23(×2), rrn4.5(×2), rrn5(×2) |
| Transfer RNAs | atrnA-UGC(×2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU,atrnG-GCC, trnG-UCC, trnH-CAU, trnH-GUG, trnI-CAU,atrnI-GAU(×2), trnK-UUU, trnL-CAA(×2), atrnLUAA, trnL-UAG, trnM-CAU, trnN-GUU(×2), trnP-UGG, trnQ-UUG, trnR-ACG(×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC(×2), atrnV-UAC, trnW-CCA, trnY-GUA |
| Photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT |
| Cytochrome | petN, petA, petL, petG,apetB,apetD |
| ATP synthase | atpA,aatpF, atpH, atpI, atpE, atpB |
| Rubisco | rbcL |
| NADH dehydrogenase | ndhJ, ndhK, ndhC,andhB(×2), ndhF, ndhD, ndhE, ndhG, ndhI,andhA, ndhH |
| ATP-dependent protease subunit P | bclpP |
| Chloroplast translational initiation factor | infA |
| Chloroplast envelope membrane protein | cemA |
| Large units | rpl33, rpl20, rpl36, rpl14,arpl16,arpl2(×2), rpl23(×2), rpl32 |
| Small units | arps16, rps2, rps14, rps4, rps18,brps12(×2), rps11, rps8,Ψ rps19, rps3, rps7(×2), rps15 |
| RNA polymerase | rpoC2,arpoC1, rpoB, rpoA |
| Miscellaneous proteins | matK, accD, ccsA |
| Hypothetical proteins and conserved reading frame | bycf3, ycf4, ycf2(×2),Ψ ycf1, ycf15(×2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Gao, J.; Feng, J.; Yang, Z.; Qi, Z.; Li, P.; Fu, C. Comparative and Phylogenetic Analyses of Complete Chloroplast Genomes of Scrophularia incisa Complex (Scrophulariaceae). Genes 2022, 13, 1691. https://doi.org/10.3390/genes13101691
Wang R, Gao J, Feng J, Yang Z, Qi Z, Li P, Fu C. Comparative and Phylogenetic Analyses of Complete Chloroplast Genomes of Scrophularia incisa Complex (Scrophulariaceae). Genes. 2022; 13(10):1691. https://doi.org/10.3390/genes13101691
Chicago/Turabian StyleWang, Ruihong, Jing Gao, Jieying Feng, Zhaoping Yang, Zhechen Qi, Pan Li, and Chengxin Fu. 2022. "Comparative and Phylogenetic Analyses of Complete Chloroplast Genomes of Scrophularia incisa Complex (Scrophulariaceae)" Genes 13, no. 10: 1691. https://doi.org/10.3390/genes13101691