
Bardet–Biedl Syndrome—Multiple Kaleidoscope Images: Insight into Mechanisms of Genotype–Phenotype Correlations


Abstract
:1. Introduction
2. Pleiotropy and Variable Expressivity in Bardet–Biedl Syndrome
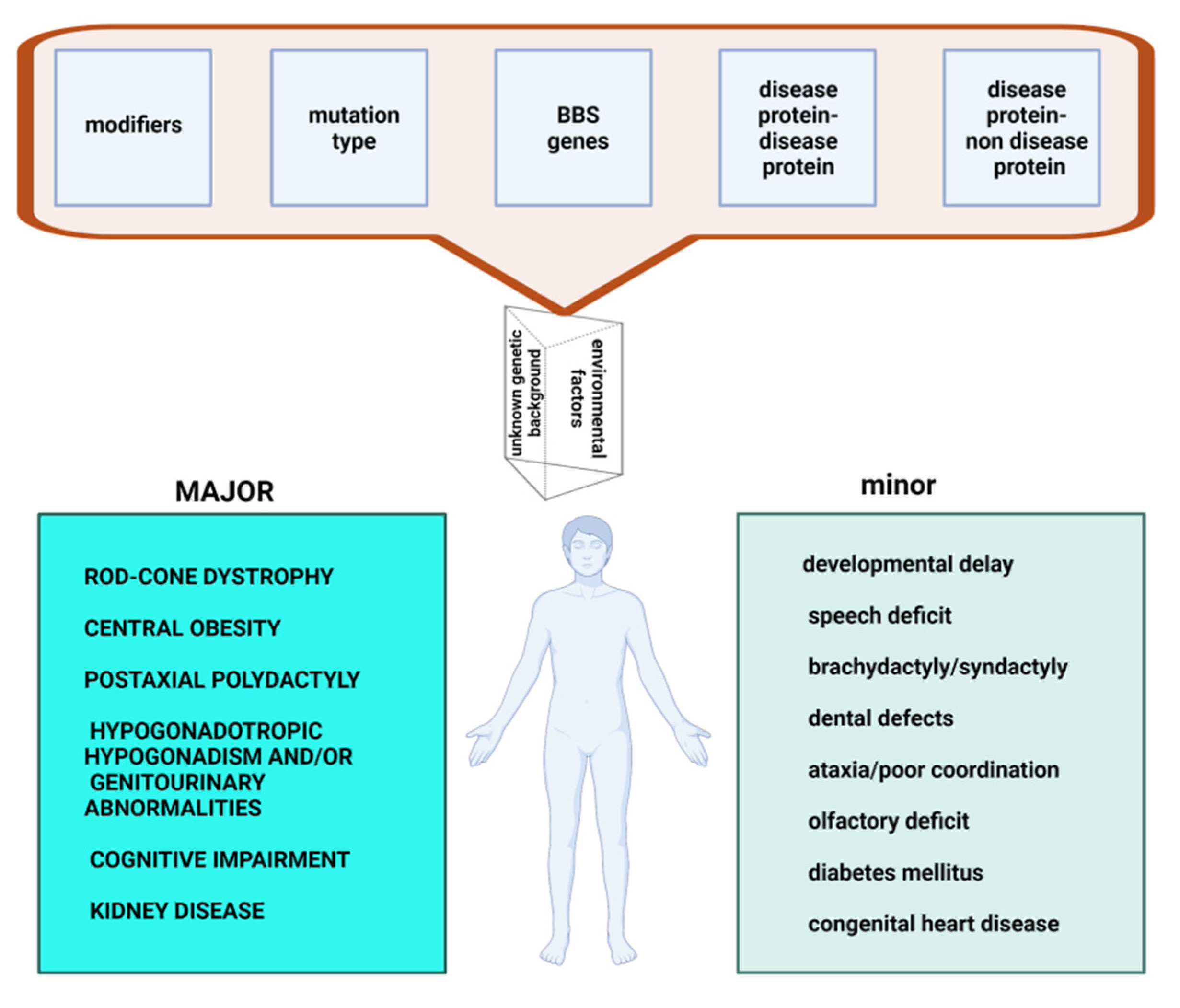
2.1. Major Features
2.2. Minor Features
3. Determinants of Clinical Effects
3.1. Locus Heterogeneity
3.2. Mutational Heterogeneity
3.3. Modifiers
3.4. Disease Protein–Non-Disease Protein Interconnectivity
3.5. Disease Protein-Disease Protein Interconnectivity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Strong, A.; Li, D.; Mentch, F.; Bedoukian, E.; Hartung, E.A.; Meyers, K.; Skraban, C.; Wen, J.; Medne, L.; Glessner, J.; et al. Ciliopathies: Coloring outside of the lines. Am. J. Med. Genet. Part A 2021, 185, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Rodriguez, M.; Badano, J.L. Ciliary biology: Understanding the cellular and genetic basis of human ciliopathies. Am. J. Med. Genet. Part C Semin. Med. Genet. 2009, 151C, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.T.; Morthorst, S.; Mogensen, J.B.; Pedersen, L.B. Primary cilia and coordination of receptor tyrosine kinase (RTK) and transforming growth factor β (TGF-β) signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez-Satta, M.; Cástro-Sánchez, S.; Valverde, D. Bardet-Biedl syndrome as a chaperonopathy: Dissecting the major role of chaperonin-like BBS proteins (BBS6-BBS10-BBS12). Front. Mol. Biosci. 2017, 4, 55. [Google Scholar] [CrossRef] [Green Version]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the Primary Cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Singla, V. The primary cilium as the cell’s antenna: Signaling at a sensory organelle. Science 2006, 313, 629–633. [Google Scholar] [CrossRef] [Green Version]
- Adamiok-Ostrowska, A.; Piekiełko-Witkowska, A. Ciliary genes in renal cystic diseases. Cells 2020, 9, 907. [Google Scholar] [CrossRef] [Green Version]
- Mitchison, H.M.; Valente, E.M. Motile and non-motile cilia in human pathology: From function to phenotypes. J. Pathol. 2017, 241, 294–309. [Google Scholar] [CrossRef]
- Ma, M. Cilia and polycystic kidney disease. Semin. Cell Dev. Biol. 2021, 110, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.S.; Sattar, S.; Massoudi, R.A.; Gleeson, J.G. Co-occurrence of distinct ciliopathy diseases in single families suggests genetic modifiers. Am. J. Med. Genet. Part A 2011, 155, 3042–3049. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Prevalence and Incidence of Rare Diseases: Bibliographic Data. Orphanet Report Series Number 1 January 2020. Available online: https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf (accessed on 5 November 2020).
- Khan, S.; Muhammad, N.; Khan, M.A.; Kamal, A.; Rehman, Z.U.; Alam Khan, S. Genetics of human Bardet-Biedl syndrome, an updates. Clin. Genet. 2016, 90, 3–15. [Google Scholar] [CrossRef]
- Ajmal, M.; Khan, M.I.; Neveling, K.; Tayyab, A.; Jaffar, S.; Sadeque, A.; Ayub, H.; Abbasi, N.M.; Riaz, M.; Micheal, S.; et al. Exome sequencing identifies a novel and a recurrent BBS1 mutation in Pakistani families with Bardet–Biedl syn-drome. Mol. Vis. 2013, 19, 644–653. [Google Scholar] [PubMed]
- Farag, T.I.; Teebi, A.S. High incidence of Bardet Biedl syndrome among the Bedouin. Clin. Genet. 2008, 36, 463–464. [Google Scholar] [CrossRef]
- Hjortshøj, T.D.; Grønskov, K.; Brondum-Nielsen, K.; Rosenberg, T. A novel founder BBS1 mutation explains a unique high prevalence of Bardet-Biedl syndrome in the Faroe Islands. Br. J. Ophthalmol. 2008, 93, 409–413. [Google Scholar] [CrossRef]
- Sheffield, V.C.; Carml, R.; Kwltek-Black, A.; Rokhlina, T.; Nishlmura, D.; Duyk, G.M.; Elbedour, K.; Sunden, S.L.; Stone, E.M. Identification of a Bardet-Biedl syndrome locus on chromosome 3 and evaluation of an efficient approach to homozygosity mapping. Hum. Mol. Genet. 1994, 3, 1331–1335. [Google Scholar] [CrossRef]
- Beales, P.L.; Elcioglu, N.; Woolf, A.; Parker, D.; Flinter, F. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J. Med. Genet. 1999, 36, 437–446. [Google Scholar]
- Sheffield, V.C.; Zhang, Q.; Heon, E.; Drack, A.V.; Stone, E.M.; Carmi, R. The Bardet-Biedl syndrome. Epstein’s Inborn Errors Dev. 2016, 237–240. [Google Scholar] [CrossRef]
- Forsythe, E.; Beales, P.L. Bardet–Biedl syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef]
- Denniston, A.K.; Beales, P.L.; Tomlins, P.J.; Good, P.; Langford, M.; Foggensteiner, L.; Williams, D.; Tsaloumas, M.D. Evaluation of visual function and needs in adult patients with Bardet–Biedl syndrome. Retina 2014, 34, 2282–2289. [Google Scholar] [CrossRef] [PubMed]
- Niederlova, V.; Modrak, M.; Tsyklauri, O.; Huranova, M.; Stepanek, O. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum. Mutat. 2019, 40, 2068–2087. [Google Scholar] [CrossRef]
- Weihbrecht, K.; Goar, W.A.; Pak, T.; Garrison, J.E.; DeLuca, A.P.; Stone, E.M.; Scheetz, T.E.; Sheffield, V.C. Keeping an eye on Bardet-Biedl syndrome: A comprehensive review of the role of Bardet-Biedl syndrome genes in the eye. Med. Res. Arch. 2017, 5, 1–21. [Google Scholar] [CrossRef]
- Mockel, A.; Perdomo, Y.; Stutzmann, F.; Letsch, J.; Marion, V.; Dollfus, H. Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies. Prog. Retin. Eye Res. 2011, 30, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.-F.; Rahmouni, K. Molecular basis of the obesity associated with Bardet–Biedl syndrome. Trends Endocrinol. Metab. 2011, 22, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Benzinou, M.; Walley, A.; Lobbens, S.; Charles, M.A.; Jouret, B.; Fumeron, F.; Balkau, B.; Meyre, D.; Froguel, P. Bardet-Biedl syndrome gene variants are associated with both childhood and adult common obesity in French Caucasians. Diabetes 2006, 55, 2876–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, E.; Lauer, J.; Durand, M.; Pelletier, V.; Obringer, C.; Claussmann, A.; Braun, J.-J.; Redin, C.; Mathis, C.; Muller, J.; et al. Mesoaxial polydactyly is a major feature in Bardet-Biedl syndrome patients withLZTFL1(BBS17) mutations. Clin. Genet. 2014, 85, 476–481. [Google Scholar] [CrossRef]
- Mujahid, S.; Hunt, K.F.; Cheah, Y.S.; Forsythe, E.; Hazlehurst, J.M.; Sparks, K.; Mohammed, S.; Tomlinson, J.A.; Amiel, S.; Carroll, P.V.; et al. The endocrine and metabolic characteristics of a large Bardet-Biedl syndrome clinic population. J. Clin. Endocrinol. Metab. 2018, 103, 1834–1841. [Google Scholar] [CrossRef]
- Forsythe, E.; Sparks, K.; Best, S.; Borrows, S.; Hoskins, B.; Sabir, A.; Barrett, T.; Williams, D.; Mohammed, S.; Goldsmith, D.; et al. Risk factors for severe renal disease in Bardet–Biedl syndrome. J. Am. Soc. Nephrol. 2016, 28, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Haws, R.M.; Joshi, A.; Shah, S.A.; Alkandari, O.; Turman, M.A. Renal transplantation in Bardet–Biedl Syndrome. Pediatr. Nephrol. 2016, 31, 2153–2161. [Google Scholar] [CrossRef]
- Braun, J.; Noblet, V.; Kremer, S.; Molière, S.; Dollfus, H.; Marion, V.; Goetz, N.; Muller, J.; Riehm, S. Value of MRI Olfactory Bulb evaluation in the assessment of olfactory dysfunction in Bardet Biedl syndrome. Clin. Genet. 2016, 90, 79–83. [Google Scholar] [CrossRef]
- Forsyth, R.L.; Gunay-Aygun, M. Bardet-Biedl Syndrome Overview. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993; 14 July 2003 (Updated 23 July 2020). Available online: https://www.ncbi.nlm.nih.gov/books/NBK1363/ (accessed on 27 June 2021).
- Tsyklauri, O.; Niederlova, V.; Forsythe, E.; Prasai, A.; Drobek, A.; Kasparek, P.; Sparks, K.; Trachtulec, Z.; Prochazka, J.; Sedlacek, R.; et al. Bardet–Biedl Syndrome ciliopathy is linked to altered hematopoiesis and dysregulated self-tolerance. EMBO Rep. 2021, 22, e50785. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The human gene mutation database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Qual. Life Res. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- HGNC, HUGO Gene Nomenclature Committee Home Page. Available online: http://www.genenames.org/ (accessed on 1 August 2012).
- Proteinatlas the Human Protein Atlas. Available online: https://www.proteinatlas.org (accessed on 19 December 2019).
- UniProt. Available online: https://www.uniprot.org/ (accessed on 26 April 2021).
- Covic, M.; Ştefănescu, D.; Sandovici, I.; Gorduza, E.V. Genetică Medicală, 3rd ed.; Polirom Iaşi: Iasi, Romania, 2017. [Google Scholar]
- Billingsley, G.; Bin, J.; Fieggen, K.; Duncan, J.L.; Gerth-Kahlert, C.; Ogata, K.; Wodak, S.I.; Traboulsi, E.A.; Fishman, G.; Paterson, A.; et al. Mutations in chaperonin-like BBS genes are a major contributor to disease development in a multiethnic Bardet-Biedl syndrome patient population. J. Med. Genet. 2010, 47, 453–463. [Google Scholar] [CrossRef]
- Deveault, C.; Billingsley, G.; Duncan, J.L.; Bin, J.; Theal, R.; Vincent, A.; Fieggen, K.; Gerth-Kahlert, C.; Noordeh, N.; Traboulsi, E.I.; et al. BBS genotype-phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Hum. Mutat. 2011, 32, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, O.; Marion, V.; Stoetzel, C.; Durand, M.; Holder, M.; Sigaudy, S.; Sarda, P.; Hamel, C.P.; Brandt, C.; Dollfus, H.; et al. Bardet-Biedl syndrome: A study of the renal and cardiovascular phenotypes in a French cohort. Clin. J. Am. Soc. Nephrol. 2010, 6, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Yu, D.; Seo, S.; Stone, E.M.; Sheffield, V.C. Intrinsic protein-protein interaction-mediated and chaperonin-assisted sequential assembly of stable Bardet-Biedl syndrome protein complex, the BBSome. J. Biol. Chem. 2012, 287, 20625–20635. [Google Scholar] [CrossRef] [Green Version]
- Muller, J.; Stoetzel, C.; Vincent, M.C.; Leitch, C.C.; Laurier, V.; Danse, J.M.; Hellé, S.; Marion, V.; Bennouna-Greene, V.; Vicaire, S.; et al. Identification of 28 novel mutations in the Bardet–Biedl syndrome genes: The burden of private mutations in an extensively heterogeneous disease. Qual. Life Res. 2010, 127, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Hjortshøj, T.D.; Grønskov, K.; Philp, A.R.; Nishimura, D.Y.; Riise, R.; Sheffield, V.; Rosenberg, T.; Brøndum-Nielsen, K. Bardet-Biedl syndrome in Denmark-report of 13 novel sequence variations in six genes. Hum. Mutat. 2010, 31, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Satta, M.; Castro-Sánchez, S.; Pereiro, I.; Piñeiro-Gallego, T.; Baiget, M.; Ayuso, C.; Valverde, D. Overview of Bardet-Biedl syndrome in Spain: Identification of novel mutations inBBS1, BBS10 and BBS12genes. Clin. Genet. 2014, 86, 601–602. [Google Scholar] [CrossRef]
- Wheway, G.; Mitchison, H.M. Genomics England research consortium opportunities and challenges for molecular understanding of ciliopathies–The 100,000 genomes project. Front. Genet. 2019, 10, 127. [Google Scholar] [CrossRef] [Green Version]
- Davis, E.; Zhang, Q.; Liu, Q.; Diplas, B.; Davey, L.M.; Hartley, J.; Stoetzel, C.; Szymanska, K.; Ramaswami, G.; Logan, C.V.; et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011, 43, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Lindstrand, A.; Frangakis, S.; Carvalho, C.M.; Richardson, E.B.; McFadden, K.A.; Willer, J.R.; Pehlivan, D.; Liu, P.; Pediaditakis, I.L.; Sabo, A.; et al. Copy-Number variation contributes to the mutational load of Bardet-Biedl syndrome. Am. J. Hum. Genet. 2016, 99, 318–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomeroy, J.; Krentz, A.D.; Richardson, J.G.; Berg, R.L.; VanWormer, J.J.; Haws, R.M. Bardet-Biedl syndrome: Weight patterns and genetics in a rare obesity syndrome. Pediatr. Obes. 2021, 16, e12703. [Google Scholar] [CrossRef]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peränen, J.; Merdes, A.; Slusarski, D.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.; et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Nachury, M. The BBSome. Curr. Biol. 2009, 19, R472–R473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, S.; Baye, L.M.; Schulz, N.P.; Beck, J.S.; Zhang, Q.; Slusarski, D.C.; Sheffield, V.C. BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc. Natl. Acad. Sci. USA 2010, 107, 1488–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- M’Hamdi, O.; Redin, C.; Stoetzel, C.; Ouertani, I.; Chaabouni, M.; Maazoul, F.; M’Rad, R.; Mandel, J.L.; Dollfus, H.; Muller, J. Clinical and genetic characterization of Bardet-Biedl syndrome in Tunisia: Defining a strategy for molecular diagnosis. Clin. Genet. 2013, 85, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, E.; Sparks, K.; Hoskins, B.; Bagkeris, E.; McGowan, B.; Carroll, P.; Huda, M.; Mujahid, S.; Peters, C.; Barrett, T.; et al. Genetic predictors of cardiovascular morbidity in Bardet–Biedl syndrome. Clin. Genet. 2014, 87, 343–349. [Google Scholar] [CrossRef]
- PhenoModifier: A Genetic Modifier Database for Elucidating the Genetic Basis of Human Phenotypic Variation. Available online: https://www.biosino.org/PhenoModifier/index (accessed on 26 July 2021).
- Leitch, C.C.; Zaghloul, N.A.; Davis, E.; Stoetzel, C.; Diaz-Font, A.; Rix, S.; Al-Fadhel, M.; Lewis, R.A.; Eyaid, W.; Banin, E.; et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008, 40, 443–448. [Google Scholar] [CrossRef]
- Bölükbaşı, E.Y.; Mumtaz, S.; Afzal, M.; Woehlbier, U.; Malik, S.; Tolun, A. Homozygous mutation in CEP19, a gene mutated in morbid obesity, in Bardet-Biedl syndrome with predominant postaxial polydactyly. J. Med. Genet. 2018, 55, 189–197. [Google Scholar] [CrossRef]
- Badano, J.L.; Leitch, C.C.; Ansley, S.J.; May-Simera, H.; Lawson, S.; Lewis, R.A.; Beales, P.L.; Dietz, H.C.; Fisher, S.J.; Katsanis, N. Dissection of epistasis in oligogenic Bardet–Biedl syndrome. Nature 2006, 439, 326–330. [Google Scholar] [CrossRef]
- Badano, J.L.; Kim, J.C.; Hoskins, B.E.; Lewis, R.A.; Ansley, S.J.; Cutler, D.J.; Castellan, C.; Beales, P.L.; Leroux, M.R.; Katsanis, N. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 2003, 12, 1651–1659. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Chung, Y.D. Ciliary subcompartments: How are they established and what are their functions? BMB Rep. 2015, 48, 380–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, B.P.; Robertson, D.L.; Hentges, K.E. Locus heterogeneity disease genes encode proteins with high interconnectivity in the human protein interaction network. Front. Genet. 2014, 5, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiens, C.J.; Tong, Y.; Esmail, M.A.; Oh, E.; Gerdes, J.; Wang, J.; Tempel, W.; Rattner, J.B.; Katsanis, N.; Park, H.-W.; et al. Bardet-Biedl syndrome-associated small GTPase ARL6 (BBS3) functions at or near the ciliary gate and modulates Wnt signaling. J. Biol. Chem. 2010, 285, 16218–16230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, T.J.; Mitchell, B.; Abitua, P.; Kintner, C.; Wallingford, J.B. Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat. Genet. 2008, 40, 871–879. [Google Scholar] [CrossRef]
- Adams, M.; Simms, R.J.; Abdelhamed, Z.; Dawe, H.R.; Szymanska, K.; Logan, C.; Wheway, G.; Pitt, E.; Gull, K.; Knowles, M.A.; et al. A meckelin–filamin A interaction mediates ciliogenesis. Hum. Mol. Genet. 2011, 21, 1272–1286. [Google Scholar] [CrossRef]
- Han, Y.M.; Kang, G.M.; Byun, K.; Ko, H.W.; Kim, J.; Shin, M.-S.; Kim, H.-K.; Gil, S.Y.; Yu, J.H.; Lee, B.; et al. Leptin-promoted cilia assembly is critical for normal energy balance. J. Clin. Investig. 2014, 124, 2193–2197. [Google Scholar] [CrossRef] [Green Version]
- Mariman, E.C.M.; Vink, R.G.; Roumans, N.J.T.; Bouwman, F.G.; Stumpel, C.T.R.M.; Aller, E.E.J.G.; van Baak, M.; Wang, P. The cilium: A cellular antenna with an influence on obesity risk. Br. J. Nutr. 2016, 116, 576–592. [Google Scholar] [CrossRef] [Green Version]
- Nager, A.R.; Goldstein, J.S.; Herranz-Pérez, V.; Portran, D.; Ye, F.; Garcia-Verdugo, J.M.; Nachury, M.V. An actin network dispatches ciliary gpcrs into extracellular vesicles to modulate signaling. Cell 2017, 168, 252–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorentzen, E.; Kikkawa, M. Decision letter: Structure and activation mechanism of the BBSome membrane protein trafficking complex. eLife 2019, 9, e53322. [Google Scholar] [CrossRef]
- Klink, B.U.; Gatsogiannis, C.; Hofnagel, O.; Wittinghofer, A.; Raunser, S. Author response: Structure of the human BBSome core complex. eLife 2020, 9, e53910. [Google Scholar] [CrossRef] [PubMed]
- Kousi, M.; Söylemez, O.; Ozanturk, A.; Mourtzi, N.; Akle, S.; Jungreis, I.; Muller, J.; Cassa, C.A.; Brand, H.; Mokry, J.A.; et al. Evidence for secondary-variant genetic burden and non-random distribution across biological modules in a recessive ciliopathy. Nat. Genet. 2020, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| No | Gene Symbol | Gene Name | Gene Groups | Chromosome | Protein | Localization | Tissue Specificity |
|---|---|---|---|---|---|---|---|
| 1 | BBS1 | Bardet–Biedl syndrome 1 | BBSome | 11q13.2 | Bardet–Biedl syndrome 1 protein | Basal body, cilium | Low tissue specificity |
| 2 | BBS2 | Bardet–Biedl syndrome 2 | BBSome | 16q13 | Bardet–Biedl syndrome 2 protein | Basal body, cilium | Low tissue specificity |
| 3 | ARL6 | ADP ribosylation factor like GTPase 6 | ARF GTPase family | 3q11.2 | ADP-ribosylation factor-like protein 6 | Basal body, cilium, cytosol, transition zone | Low tissue specificity |
| 4 | BBS4 | Bardet–Biedl syndrome 4 | Tetratricopeptide repeat domain containing BBSome | 15q24.1 | Bardet–Biedl syndrome 4 protein | Basal body, cilium | Low tissue specificity |
| 5 | BBS5 | Bardet–Biedl syndrome 5 | BBSome | 2q31.1 | Bardet–Biedl syndrome 5 protein | Basal body | Low tissue specificity |
| 6 | MKKS | MKKS centrosomal shuttling protein | Chaperonins | 20p12.2 | McKusick–Kaufman/Bardet–Biedl syndromes putative chaperonin | Basal body, cytosol | Low tissue specificity |
| 7 | BBS7 | Bardet–Biedl syndrome 7 | BBSome | 4q27 | Bardet–Biedl syndrome 7 protein | Basal body, cilium | Low tissue specificity |
| 8 | TTC8 | Tetratricopeptide repeat domain 8 | Tetratricopeptide repeat domain containing BBSome | 14q31.3 | Tetratricopeptide repeat protein 8 | Basal body, cilium, IFT | Low tissue specificity |
| 9 | BBS9 | Bardet–Biedl syndrome 9 | BBSome | 7p14.3 | Protein PTHB1 | Cilium | Low tissue specificity |
| 10 | BBS10 | Bardet–Biedl syndrome 10 | Chaperonins | 12q21.2 | Bardet–Biedl syndrome 10 protein | Basal body | Low tissue specificity |
| 11 | TRIM32 | Tripartite motif containing 32 | Tripartite motif containing Ring finger proteins | 9q33.1 | E3 ubiquitin-protein ligase TRIM32 | Intermediate filaments | Low tissue specificity |
| 12 | BBS12 | Bardet–Biedl syndrome 12 | Chaperonins | 4q27 | Bardet–Biedl syndrome 12 protein | Basal body | Low tissue specificity |
| 13 | MKS1 | MKS transition zone complex subunit 1 | B9 domain containing MKS complex | 17q22 | Meckel syndrome type 1 protein | Basal body | Low tissue specificity |
| 14 | CEP290 | centrosomal protein 290 | MKS complex | 12q21.32 | Centrosomal protein of 290 kDa (Cep290) | Basal body, centrosome | Low tissue specificity |
| 15 | WDPCP | WD repeat containing planar cell polarity effector | Ciliogenesis and planar polarity effector complex subunits | 2p15 | WD repeat-containing and planar cell polarity effector protein fritz homolog (hFRTZ) | Cytosol, plasma membrane, axoneme | Low tissue specificity |
| 16 | SDCCAG8 | SHH signaling and ciliogenesis regulator SDCCAG8 | MicroRNA protein coding host genes | 1q43-q44 | Serologically defined colon cancer antigen 8 | Basal body, centriole, transition zone | Low tissue specificity |
| 17 | LZTFL1 | leucine zipper transcription factor like 1 | 3p21.31 | Leucine zipper transcription factor-like protein 1 | Basal body, cilium | Tissue enhanced (lymphoid tissue) | |
| 18 | BBIP1 | BBSome interacting protein 1 | BBSome | 10q25.2 | BBSome-interacting protein 1 | Cytoplasm, cytosol | Tissue enhanced (testis) |
| 19 | IFT27 | intraflagellar transport 27 | IFT-B1 complex RAB, member RAS oncogene GTPases | 22q12.3 | Intraflagellar transport protein 27 homolog | Basal body, cilium, IFT | Low tissue specificity |
| 20 | IFT74 | intraflagellar transport 74 | IFT-B1 complex | 9p21.2 | Intraflagellar transport protein 74 homolog | Basal body, cilium, IFT | Low tissue specificity |
| 21 | C8orf37 | chromosome 8 open reading frame 37 | 8q22.1 | Protein C8orf37 | Basal body, ciliary root | Low tissue specificity | |
| 22 | SCLT1 | sodium channel and clathrin linker 1 | 4q28.2 | Sodium channel and clathrin linker 1 | Centriole | Low tissue specificity | |
| 23 | NPHP1 | Nephrocystin 1 | NPHP complex | 2q13 | Nephrocystin-1 | Transition zone | Tissue enhanced (skeletal muscle) |
| 24 | SCAPER | S-phase cyclin A associated protein in the ER | Zinc fingers C2H2-type | 15q24.3 | S phase cyclin A-associated protein in the endoplasmic reticulum (S phase cyclin A-associated protein in the ER) | Endoplasmic reticulum | Low tissue specificity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florea, L.; Caba, L.; Gorduza, E.V. Bardet–Biedl Syndrome—Multiple Kaleidoscope Images: Insight into Mechanisms of Genotype–Phenotype Correlations. Genes 2021, 12, 1353. https://doi.org/10.3390/genes12091353
Florea L, Caba L, Gorduza EV. Bardet–Biedl Syndrome—Multiple Kaleidoscope Images: Insight into Mechanisms of Genotype–Phenotype Correlations. Genes. 2021; 12(9):1353. https://doi.org/10.3390/genes12091353
Chicago/Turabian StyleFlorea, Laura, Lavinia Caba, and Eusebiu Vlad Gorduza. 2021. "Bardet–Biedl Syndrome—Multiple Kaleidoscope Images: Insight into Mechanisms of Genotype–Phenotype Correlations" Genes 12, no. 9: 1353. https://doi.org/10.3390/genes12091353