
ATM-Mediated Mitochondrial Radiation Responses of Human Fibroblasts


Department of Environmental Health, National Institute of Public Health 2-3-6 Minami, Wako 351-0197, Saitama, Japan
Genes 2021, 12(7), 1015; https://doi.org/10.3390/genes12071015
Submission received: 25 May 2021
/
Revised: 25 June 2021
/
Accepted: 29 June 2021
/
Published: 30 June 2021
(This article belongs to the Special Issue Role of ATM and MRE11 in Genomic Stability and Oxidative Stress Responses)
{kind=link}
{kind=link}
Abstract
:Ataxia telangiectasia (AT) is characterized by extreme sensitivity to ionizing radiation. The gene mutated in AT, Ataxia Telangiectasia Mutated (ATM), has serine/threonine protein kinase activity and mediates the activation of multiple signal transduction pathways involved in the processing of DNA double-strand breaks. Reactive oxygen species (ROS) created as a byproduct of the mitochondria’s oxidative phosphorylation (OXPHOS) has been proposed to be the source of intracellular ROS. Mitochondria are uniquely vulnerable to ROS because they are the sites of ROS generation. ROS-induced mitochondrial mutations lead to impaired mitochondrial respiration and further increase the likelihood of ROS generation, establishing a vicious cycle of further ROS production and mitochondrial damage. AT patients and ATM-deficient mice display intrinsic mitochondrial dysfunction and exhibit constitutive elevations in ROS levels. ATM plays a critical role in maintaining cellular redox homeostasis. However, the precise mechanism of ATM-mediated mitochondrial antioxidants remains unclear. The aim of this review paper is to introduce our current research surrounding the role of ATM on maintaining cellular redox control in human fibroblasts. ATM-mediated signal transduction is important in the mitochondrial radiation response. Perturbation of mitochondrial redox control elevates ROS which are key mediators in the development of cancer by many mechanisms, including ROS-mediated genomic instability, tumor microenvironment formation, and chronic inflammation.
1. Introduction
Ataxia telangiectasia (AT) is a rare autosomal recessive disorder characterized by progressive impairments in muscular coordination, immunodeficiency, radiosensitivity, and a predisposition to cancer. The overall prevalence of AT is about 1:100,000 live births in Japan. The gene that is mutated in AT, Ataxia Telangiectasia Mutated (ATM) and ATM- and Rad3-related (ATR), encodes large serine/threonine protein kinases that orchestrate nuclear DNA damage responses (DDR) with a multitude of substrates. ATM and ATR have distinct DNA damage specificities, and their functions are not identical. Double-strand breaks (DSB) can arise from metabolic processes, DNA replication stress, and DNA-damaging agents such as ionizing radiation, UV light, and genotoxic molecules. Recognizing DNA damage is the critical first step of the DDR. The trimeric MRE11–RAD50–NBS1 (MRN) complex is involved in DSB recognition, pre-repair mechanisms, and keeping sister chromatids or broken ends in close proximity to one another [1]. This complex recruits ATM to the DSB site [2], which, in turn, initiates DNA repair and checkpoint responses by activating an extensive signaling network [3,4,5,6]. ATM is involved in maintaining genomic integrity and defends against endogenous as well as exogenous DNA damage [7]. ATM also has a role as a sensor of ROS and is activated by treatment with H2O2 [8]. Loss of ATM function leads to nuclear genomic instability and reactive oxygen species (ROS)-mediated oxidative stress, which is thought to be a causal factor in the development of AT [5,6,7,9,10,11]. Elevated oxidative stress and mitochondrial abnormalities have been reported in AT patients and in ATM-deficient mice [10,12,13,14]. Antioxidants can prevent intrinsic defects and cancer susceptibility in ATM-deficient mice [15,16]. However, how ATM is involved in cellular redox homeostasis remains unclear. This article will review current research regarding the role of ATM in radiation-induced oxidative stress, in which the mitochondria play a critical role. Mitochondrial ROS are associated with radiation-related cancers. An analysis of radiation-induced oxidative stress is vital to estimate radiation cancer risks.
2. Mitochondrial ROS Generation and Redox Control
ROS are highly reactive chemical ions such as superoxide anions (O2−), hydroxyl radical (OH−), peroxy radical (RO2−), hydrogen peroxide (H2O2), and reactive nitrogen species. Mitochondria are central to energy production, generating adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS). Endogenous ROS are generated during necessary metabolic reactions in the mitochondria of eukaryotic cells. Mitochondrial DNA (mtDNA) is located near the site of ROS production (Lambert and Brand, 2009) and is therefore especially vulnerable to ROS-induced damage [17]. In turn, this makes genes encoded by mtDNA especially sensitive to oxidative-stress-induced mutations (Wallace, 2010). Complexes I and III in the mitochondria are the sites of ROS production and electron leak from the electron transport chain reaction in the form of O2− [18,19,20]. The nicotinamide adenine dinucleotide phosphate oxidative enzymes (NOXs) produce ROS in phagocytes and in other tissues [21]. ROS attack a large number of biomolecules such as proteins, lipids, carbohydrates, and nucleic acids. H2O2 is less reactive than O2−; it can also diffuse across the mitochondria to mediate oxidative signaling [22].
The cellular redox balance is regulated by eliminating ROS with antioxidants. Molecules involved in the mitochondrial antioxidant defense system such as glutathione (GSH) are required for maintaining intracellular redox homeostasis [23]. Manganese superoxide dismutase (MnSOD) and glutathione peroxidase (GPx) are responsible for removing ROS. MnSOD catalyzes the dismutation of O2− into H2O2 and O2. GPx is a scavenger of H2O2 that converts GSH into oxidized glutathione (glutathione disulfide, GSSG) and simultaneously reduces H2O2 into H2O [23,24,25]. GSH is regenerated from GSSG using NADPH-dependent glutathione reductase. The GSH/GSSG ratio is thought to be a useful indicator of oxidative stress within cells. Further, disruption of cellular redox homeostasis leads to ROS-mediated oxidative stress which causes detrimental health effects, including cancer, neurodegenerative diseases, and cardiac diseases [26,27,28,29,30].
3. Radiation-Induced Oxidative Stress
Radiation induces oxidative stress in cells [31]. Radiation primarily generates ROS via the radiolysis of water. Subsequently, the delayed production of mitochondrial ROS mediates the long-lasting effects of radiation [32]. Cellular antioxidant defense systems maintain ROS levels after exposure to low-to-moderate doses of ionizing radiation. Acute single radiation (SR) of less than 2 Gy transiently increases ROS concentrations, which subsequently return to baseline levels 24 h after irradiation, in human fibroblasts [32,33]. In contrast, high doses of radiation (more than 5 Gy) are associated with the induction of oxidative stress, suggesting that the antioxidant defense system is ineffective in this scenario. The GSH reaction is critical in maintaining redox homeostasis following radiation. The effect of radiation on GSH redox control was investigated in human fibroblasts (manuscript submitted). Although reduced GSH was present in the irradiated cells, radiation inactivated GPx. Different radiation exposure methods were used to evaluate the effects of fractionated radiation (FR) on radiation-induced oxidative stress. Clinical radiotherapy is given by dividing a dose of radiation into multiple fractions over several weeks [34]. Repeated radiation exposures in low doses (0.01 Gy or 0.05 Gy per fraction) for 21 days (total dose of 0.3 Gy or 1.5 Gy) induced prolonged ROS generation, leading to chronic oxidative stress in FR-treated cells [35]. Low doses of FR are equivalent to high doses of SR on mitochondrial ROS induction. Similarly to SR, FR in low doses causes downregulation of GPx activity. Thus, a decrease in GPx activity is an important contributor to radiation-induced oxidative stress. Essentially, radiation increases mitochondrial ROS levels according to the radiation dose and irradiation conditions in human fibroblasts [26].
4. Mitochondrial Radiation Responses
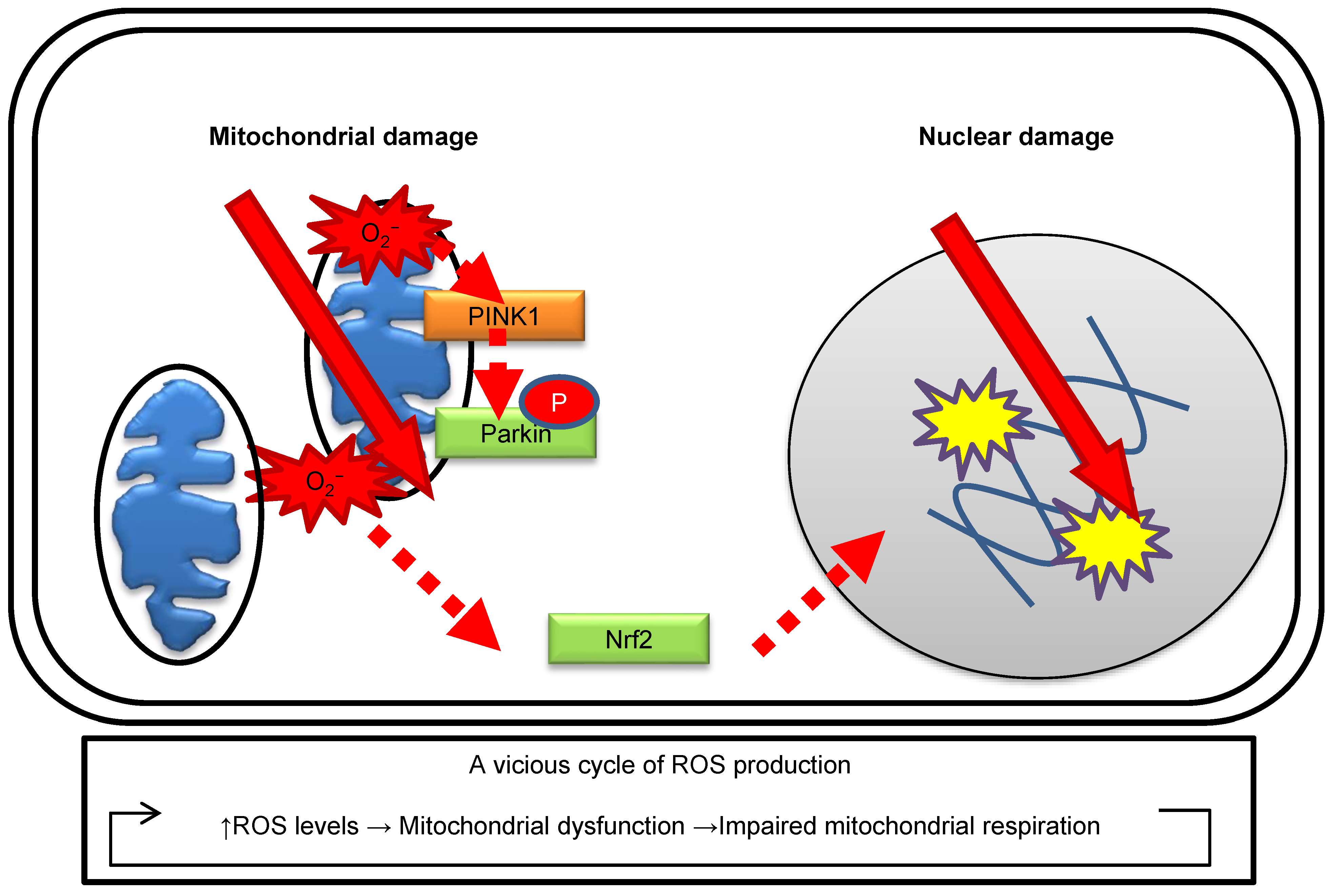
The role of mitochondria in the radiation response is well summarized in other mitochondrial biology papers [36]. The effects of ionizing radiation on the nuclei and mitochondria are illustrated in Figure 1. Radiation increases mitochondrial ROS levels according to the radiation dose and irradiation conditions in human fibroblasts [32]. Radiation-induced oxidative damage of mtDNA can be detected as increases in 8-hydroxydeoxyguanosine (8-OHdG) levels via high-performance liquid chromatography–electrochemical detectors (HPLC–ECD) [35]. Radiation activates mitochondrial biogenesis to aid in the increased energy demands for DDR [35,37]. As described above, mitochondria have a role in ROS generation during OXPHOS under irradiation. Radiation damage to the mitochondria leads to impairments in mitochondrial respiration and makes them more prone to ROS generation [36,38]. This leads to a vicious cycle of further ROS production and mitochondrial DNA damage. The selective removal of damaged mitochondria, known as mitophagy, is indispensable to cellular survival in cases where damaged mitochondria are releasing cytotoxic materials. In response to a loss of mitochondrial membrane potential, phosphatase and tensin homolog induced putative kinase 1 (PINK1), a mitochondrial kinase, phosphorylates Parkin, an E3-like ubiquitin-ligase, which, in turn, localizes specifically to impaired mitochondria [39]. Radiation-induced mitochondrial ROS are associated with the induction of mitophagy, which can be detected by the formation of Parkin and activation of the antioxidative response of nuclear factor erythroid 2-related factor 2 (Nrf2) in human fibroblasts [32,35]. Parkin can be utilized as a marker to identify oxidative damage in mitochondria.
Mitochondria undergo morphological changes in order to maintain healthy mitochondrial networks by a balance of fission and fusion events [40,41]. Following radiation exposure, the mitochondrial morphology is remarkably altered to maintain its function. Such mitochondrial dynamics occur via the fusion of healthy mitochondria with the damaged ones [42,43,44]. In fact, low-dose radiation stimulates mitochondrial fusion in order to protect rat neurons [45]. On the other hand, a high dose of radiation induces dynamin-related protein 1 (Drp1)-mediated mitochondrial fission in normal human fibroblast-like cells [46]. Mitochondria are also associated with radiation-induced adaptive responses, the mitochondrial bystander effect, and genomic instability. MnSOD exists within mitochondria and promotes adaptive radiation responses as a signaling regulator of cell survival pathways [47]. ROS activates NF-kappaB signaling pathways, which are implicated in the regulation of the radiation-induced bystander effect [48]. Intercellular communication is mediated by mitochondrial transfer from one cell to another under stress conditions [49]. Mitochondria are key players in activating apoptosis in mammalian cells. In apoptosis, cytochrome c is released from the mitochondria by the permeability transition pore and subsequently activates apoptosis signaling pathways. [50]. Collectively, mitochondria are an important radiation target.
A part of ATM is located in the mitochondria and responds to mitochondrial dysfunction [51]. Radiation activates ATM, which protects mitochondrial quality [32,52,53]. ATM has a role in inducing mitophagy after selective elimination of damaged mitochondrial components by mitochondrial fission. Additionally, ATM loss leads to severe radiation-induced mitochondrial damage in human fibroblasts [52]. ATM is frequently mutated in cancers. Cells with ATM mutation may display a highly radiosensitive phenotype with a lack of the normal ATM-mediated mitochondrial damage response including OXPHOS activation, ROS generation, and mitophagy. ATM plays a direct role in modulating mitochondrial homeostasis under genotoxic stress.
5. ATM and AMPK-Mediated Interconnected Damage Signaling Networks between the Nuclei and Mitochondria
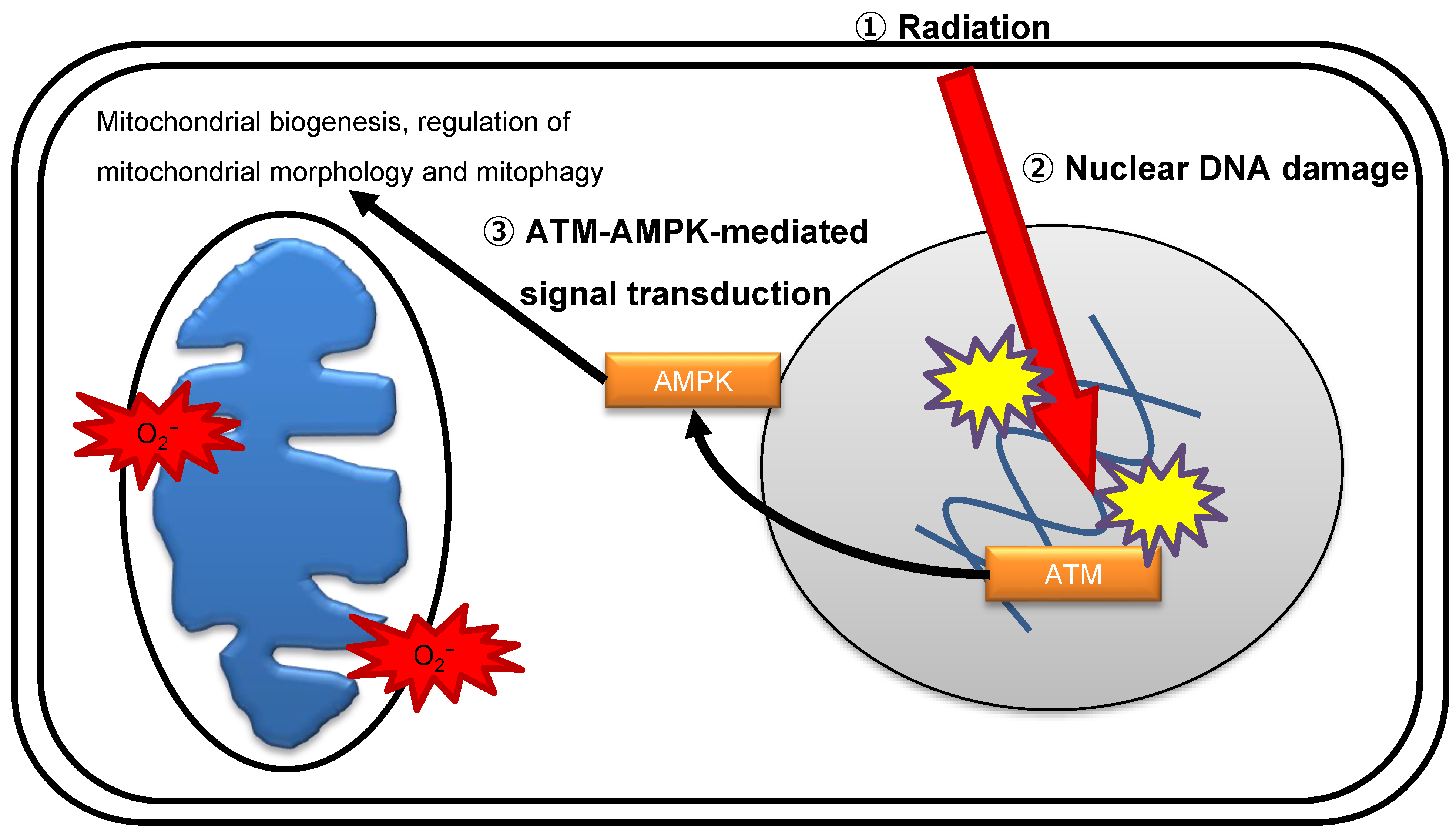
Radiation immediately stimulates multiple comprehensive molecular signaling pathways to execute DDR. ATM is considered the master sensor of DSBs and transduces the DNA damage signal to trigger the activity of downstream effectors by biochemical modification reactions [54]. The nuclear DNA damage signal diffuses to other intracellular organelles, including mitochondria, in irradiated cells. AMP-activated protein kinase (AMPK) protects cells against physiological stress by sensing energy stress when the ATP:AMP/ADP ratio declines and suppresses cell growth via regulating the mammalian target of rapamycin (mTOR) pathway [55]. AMPK controls various aspects of mitochondrial homeostasis by promoting mitochondrial biogenesis, regulation of mitochondrial morphology, and mitophagy [56]. One study suggests that ATM is an upstream kinase for the activation of AMPK by phosphorylation on Thr172 [57], while another study suggests that ATM does not directly phosphorylate AMPK [58]. The precise mechanism of ATM-mediated phosphorylation of AMPK after irradiation is under current investigation. ATM controls mitochondrial quality following radiation exposure [32,52,53]. AMPK acts as a mediator of DNA damage signals to mitochondria in response to genomic stress. DNA damage activates the ATM–AMPK–peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) signaling pathway, which facilitates mitochondrial biogenesis (Figure 2) [52,57]. ROS released from mitochondria cause indirect oxidative damage to nuclear DNA, which subsequently induces long-lasting oxidative damage, in irradiated cells. AMPK activates p53 and cyclin-dependent kinase inhibitors p21-dependent cell cycle arrest [59]. Collectively, ATM and AMPK are key molecules in the crosstalk between the nucleus and mitochondria under genomic stress conditions inflicted by ionizing radiation.
6. The Role of Oxidative Stress in Cancer
High ROS concentrations in cancerous cells have been proposed to arise from mitochondrial dysfunction [60]. Mitochondrial genomic instability is associated with increased incidence of various diseases, including metabolic diseases, neurodegenerative diseases, and cancer [27,61,62,63]. Oxidants have genotoxic effects and can promote the development of multistage carcinogenesis [64]. Indeed, mtDNA mutations have been reported in various types of tumors [65,66,67]. Impairment of mitochondrial respiration alters the cellular metabolism of cancer cells [68]. Cancer cells require a much higher glucose supply to maintain their high proliferation rate, and they prefer to metabolize glucose by glycolysis. The mitochondrial antioxidant enzyme MnSOD contributes to the protection of mitochondria against oxidative stress and plays a role in tumor prevention [69]. These results indicate a possible association between mitochondria-mediated oxidative stress and carcinogenesis [36,70]. ROS are key mediators in the development of cancer via many mechanisms, including ROS-mediated genomic instability, tumor cell proliferation, and chronic inflammation [71,72].
The tumor microenvironment has been widely implicated in tumorigenesis. Tumor microenvironments consist of stromal cells including myofibroblasts and/or cancer-associated fibroblasts (CAFs), vascular cells, and immune cells. Stromal fibroblasts in cancer, called CAF, communicate with malignant tumor cells via the release of tumor cell growth factors and play key roles in tumor initiation, progression, and metastasis. α-smooth muscle actin (α-SMA) is a known marker of CAF [73]. ROS-mediated signaling can regulate the formation of tumor microenvironments in tumorigenesis [74]. We recently revealed that radiation affects malignant cancer cells and can also cause molecular alterations in stromal fibroblasts. Radiation stimulates fibroblast activation via mitochondrial ROS-mediated transforming growth factor-β signaling [75]. Interactions between radiation-activated fibroblasts and malignant cancer cells contribute to the formation of the tumor microenvironment [75]. Mitochondrial dysfunction may therefore facilitate radiation-induced carcinogenesis.
7. Conclusions
ATM has a critical role in mitochondrial radiation responses. Cross-talk between the nuclei and mitochondria following radiation is important in maintaining cellular redox control. Mitochondrial oxidative stress is a key player in radiation-induced cancers. Further investigation is needed to clarify the risks of radiation.
Funding
This research was supported by a grant from Research project on the Health Effects of Radiation organized by Ministry of the Environment, Japan, the JSPS KAKENHI Grant Number 18H03377, Industrial Disease Clinical Research Grants from the Japanese Ministry of Health, Labour, and Welfare, and in part by the NIFS Collaborative Research Program (NIFS13KOBA028). This work was performed at the Joint Usage/Research Center (Radiation Biology Center), Kyoto University, and the Program of the network-type Joint Usage/Research Center for Radiation Disaster Medical Science of Hiroshima University, Nagasaki University, and Fukushima Medical University.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
I thank Akira Ushiyama and Naoki Kunugita for their support in this study.
Conflicts of Interest
The author declares no conflict of interest.
References
- Scott, S.P.; Pandita, T.K. The cellular control of DNA double-strand breaks. J. Cell. Biochem. 2006, 99, 1463–1475. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavin, M.F.; Shiloh, Y. The genetic defect in ataxia-telangiectasia. Annu. Rev. Immunol. 1997, 15, 177–202. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Kastan, M.B. ATM: Genome stability, neuronal development, and cancer cross paths. Adv. Cancer Res. 2001, 83, 209–254. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM: Expanding roles as a chief guardian of genome stability. Exp. Cell Res. 2014, 329, 154–161. [Google Scholar] [CrossRef]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Barzilai, A.; Rotman, G.; Shiloh, Y. ATM deficiency and oxidative stress: A new dimension of defective response to DNA damage. DNA Repair 2002, 1, 3–25. [Google Scholar] [CrossRef]
- Kamsler, A.; Daily, D.; Hochman, A.; Stern, N.; Shiloh, Y.; Rotman, G.; Barzilai, A. Increased oxidative stress in ataxia telangiectasia evidenced by alterations in redox state of brains from Atm-deficient mice. Cancer Res. 2001, 61, 1849–1854. [Google Scholar]
- Liu, N.; Stoica, G.; Yan, M.; Scofield, V.L.; Qiang, W.; Lynn, W.S.; Wong, P.K. ATM deficiency induces oxidative stress and endoplasmic reticulum stress in astrocytes. Lab. Investig. J. Tech. Methods Pathol. 2005, 85, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Barlow, C.; Dennery, P.A.; Shigenaga, M.K.; Smith, M.A.; Morrow, J.D.; Roberts, L.J., 2nd; Wynshaw-Boris, A.; Levine, R.L. Loss of the ataxia-telangiectasia gene product causes oxidative damage in target organs. Proc. Natl. Acad. Sci. USA 1999, 96, 9915–9919. [Google Scholar] [CrossRef] [Green Version]
- Reichenbach, J.; Schubert, R.; Schindler, D.; Müller, K.; Böhles, H.; Zielen, S. Elevated oxidative stress in patients with ataxia telangiectasia. Antioxid. Redox Signal. 2002, 4, 465–469. [Google Scholar] [CrossRef]
- Valentin-Vega, Y.A.; Maclean, K.H.; Tait-Mulder, J.; Milasta, S.; Steeves, M.; Dorsey, F.C.; Cleveland, J.L.; Green, D.R.; Kastan, M.B. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 2012, 119, 1490–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reliene, R.; Schiestl, R.H. Antioxidant N-acetyl cysteine reduces incidence and multiplicity of lymphoma in Atm deficient mice. DNA Repair 2006, 5, 852–859. [Google Scholar] [CrossRef]
- Schubert, R.; Erker, L.; Barlow, C.; Yakushiji, H.; Larson, D.; Russo, A.; Mitchell, J.B.; Wynshaw-Boris, A. Cancer chemoprevention by the antioxidant tempol in Atm-deficient mice. Hum. Mol. Genet. 2004, 13, 1793–1802. [Google Scholar] [CrossRef]
- Zhang, Y.; Lee, J.H.; Paull, T.T.; Gehrke, S.; D’Alessandro, A.; Dou, Q.; Gladyshev, V.N.; Schroeder, E.A.; Steyl, S.K.; Christian, B.E.; et al. Mitochondrial redox sensing by the kinase ATM maintains cellular antioxidant capacity. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef] [Green Version]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ROS generation and its regulation: Mechanisms involved in H(2)O(2) signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Marí, M.; de Gregorio, E.; de Dios, C.; Roca-Agujetas, V.; Cucarull, B.; Tutusaus, A.; Morales, A.; Colell, A. Mitochondrial glutathione: Recent insights and role in disease. Antioxidants 2020, 9, 909. [Google Scholar] [CrossRef] [PubMed]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St Clair, D.K. Manganese superoxide dismutase: Guardian of the powerhouse. Int. J. Mol. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef]
- Panfili, E.; Sandri, G.; Ernster, L. Distribution of glutathione peroxidases and glutathione reductase in rat brain mitochondria. FEBS Lett. 1991, 290, 35–37. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Lefer, D.J.; Granger, D.N. Oxidative stress and cardiac disease. Am. J. Med. 2000, 109, 315–323. [Google Scholar] [CrossRef]
- Simonian, N.A.; Coyle, J.T. Oxidative stress in neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 1996, 36, 83–106. [Google Scholar] [CrossRef]
- Zhu, X.; Su, B.; Wang, X.; Smith, M.A.; Perry, G. Causes of oxidative stress in Alzheimer disease. Cell. Mol. Life Sci. 2007, 64, 2202–2210. [Google Scholar] [CrossRef]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimura, T.; Sasatani, M.; Kawai, H.; Kamiya, K.; Kobayashi, J.; Komatsu, K.; Kunugita, N. ATM-mediated mitochondrial damage response triggered by nuclear DNA damage in normal human lung fibroblasts. Cell Cycle 2017, 16, 2345–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimura, T.; Ando, T.; Narao, M.; Sasatani, M.; Kamiya, K.; Ushiyama, A. Mechanism of turnover or persistence of radiation-induced myofibroblast in vitro. Cell Cycle 2020, 19, 3375–3385. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Tannock, I.F. Repopulation of cancer cells during therapy: An important cause of treatment failure. Nat. Rev. Cancer 2005, 5, 516–525. [Google Scholar] [CrossRef]
- Shimura, T.; Sasatani, M.; Kamiya, K.; Kawai, H.; Inaba, Y.; Kunugita, N. Mitochondrial reactive oxygen species perturb AKT/cyclin D1 cell cycle signaling via oxidative inactivation of PP2A in lowdose irradiated human fibroblasts. Oncotarget 2016, 7, 3559–3570. [Google Scholar] [CrossRef] [Green Version]
- Kam, W.W.; Banati, R.B. Effects of ionizing radiation on mitochondria. Free. Radic. Biol. Med. 2013, 65, 607–619. [Google Scholar] [CrossRef]
- Bartoletti-Stella, A.; Mariani, E.; Kurelac, I.; Maresca, A.; Caratozzolo, M.F.; Iommarini, L.; Carelli, V.; Eusebi, L.H.; Guido, A.; Cenacchi, G.; et al. γ rays induce a p53-independent mitochondrial biogenesis that is counter-regulated by HIF1α. Cell Death Dis. 2013, 4, e663. [Google Scholar] [CrossRef] [Green Version]
- Shimura, T.; Kunugita, N. Mitochondrial reactive oxygen species-mediated genomic instability in low-dose irradiated human cells through nuclear retention of cyclin D1. Cell Cycle 2016, 15, 1410–1414. [Google Scholar] [CrossRef]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Ono, T.; Isobe, K.; Nakada, K.; Hayashi, J.I. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. 2001, 28, 272–275. [Google Scholar] [CrossRef]
- Chien, L.; Chen, W.K.; Liu, S.T.; Chang, C.R.; Kao, M.C.; Chen, K.W.; Chiu, S.C.; Hsu, M.L.; Hsiang, I.C.; Chen, Y.J.; et al. Low-dose ionizing radiation induces mitochondrial fusion and increases expression of mitochondrial complexes I and III in hippocampal neurons. Oncotarget 2015, 6, 30628–30639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobashigawa, S.; Suzuki, K.; Yamashita, S. Ionizing radiation accelerates Drp1-dependent mitochondrial fission, which involves delayed mitochondrial reactive oxygen species production in normal human fibroblast-like cells. Biochem. Biophys. Res. Commun. 2011, 414, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Yan-Sanders, Y.; Lyn-Cook, B.D.; Wang, T.; Tamae, D.; Ogi, J.; Khaletskiy, A.; Li, Z.; Weydert, C.; Longmate, J.A.; et al. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Mol. Cell. Biol. 2003, 23, 2362–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech. Ageing Dev. 2018, 170, 30–36. [Google Scholar] [CrossRef]
- Jin, S.; Cordes, N. ATM controls DNA repair and mitochondria transfer between neighboring cells. Cell Commun. Signal. 2019, 17, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar] [PubMed]
- Morita, A.; Tanimoto, K.; Murakami, T.; Morinaga, T.; Hosoi, Y. Mitochondria are required for ATM activation by extranuclear oxidative stress in cultured human hepatoblastoma cell line Hep G2 cells. Biochem. Biophys. Res. Commun. 2014, 443, 1286–1290. [Google Scholar] [CrossRef]
- Shimura, T.; Kobayashi, J.; Komatsu, K.; Kunugita, N. Severe mitochondrial damage associated with low-dose radiation sensitivity in ATM- and NBS1-deficient cells. Cell Cycle 2016, 15, 1099–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanli, T.; Rashid, A.; Liu, C.; Harding, S.; Bristow, R.G.; Cutz, J.C.; Singh, G.; Wright, J.; Tsakiridis, T. Ionizing radiation activates AMP-activated kinase (AMPK): A target for radiosensitization of human cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. The ATM-mediated DNA-damage response: Taking shape. Trends Biochem. Sci. 2006, 31, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Wan, S.; Lyu, Y.L.; Liu, L.F.; Qi, H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS ONE 2008, 3, e2009. [Google Scholar] [CrossRef] [PubMed]
- Yee, S.W.; Chen, L.; Giacomini, K.M. The role of ATM in response to metformin treatment and activation of AMPK. Nat. Genet. 2012, 44, 359–360. [Google Scholar] [CrossRef]
- Sanli, T.; Steinberg, G.R.; Singh, G.; Tsakiridis, T. AMP-activated protein kinase (AMPK) beyond metabolism. Cancer Biol. Ther. 2014, 15, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.; Mercer, J.; Bennett, M. Mitochondria in vascular disease. Cardiovasc. Res. 2012, 95, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; ME, L.L. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wu, J.; Dressman, D.C.; Iacobuzio-Donahue, C.; Markowitz, S.D.; Velculescu, V.E.; Diaz, L.A., Jr.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 2010, 464, 610–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589. [Google Scholar] [CrossRef]
- Fliss, M.S.; Usadel, H.; Caballero, O.L.; Wu, L.; Buta, M.R.; Eleff, S.M.; Jen, J.; Sidransky, D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science 2000, 287, 2017–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Robbins, D.; Zhao, Y. Manganese superoxide dismutase in cancer prevention. Antioxid. Redox Signal. 2014, 20, 1628–1645. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Szumiel, I. Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: The pivotal role of mitochondria. Int. J. Radiat. Biol. 2015, 91, 1–12. [Google Scholar] [CrossRef]
- Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer. Oncology 2002, 16, 217–226, 229; discussion 230. [Google Scholar] [PubMed]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive oxygen species in the tumor microenvironment: An overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimura, T.; Sasatani, M.; Kawai, H.; Kamiya, K.; Kobayashi, J.; Komatsu, K.; Kunugita, N. Radiation-induced myofibroblasts promote tumor growth via mitochondrial ROS-activated TGFbeta signaling. Mol. Cancer Res. 2018, 16, 1676–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Effects of ionizing radiation on the nuclei and mitochondria.

Figure 2.
ATM-mediated crosstalk between the nuclei and mitochondria.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shimura, T. ATM-Mediated Mitochondrial Radiation Responses of Human Fibroblasts. Genes 2021, 12, 1015. https://doi.org/10.3390/genes12071015
AMA Style
Shimura T. ATM-Mediated Mitochondrial Radiation Responses of Human Fibroblasts. Genes. 2021; 12(7):1015. https://doi.org/10.3390/genes12071015
Chicago/Turabian StyleShimura, Tsutomu. 2021. "ATM-Mediated Mitochondrial Radiation Responses of Human Fibroblasts" Genes 12, no. 7: 1015. https://doi.org/10.3390/genes12071015
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.