
Role of Histone Methylation in Maintenance of Genome Integrity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
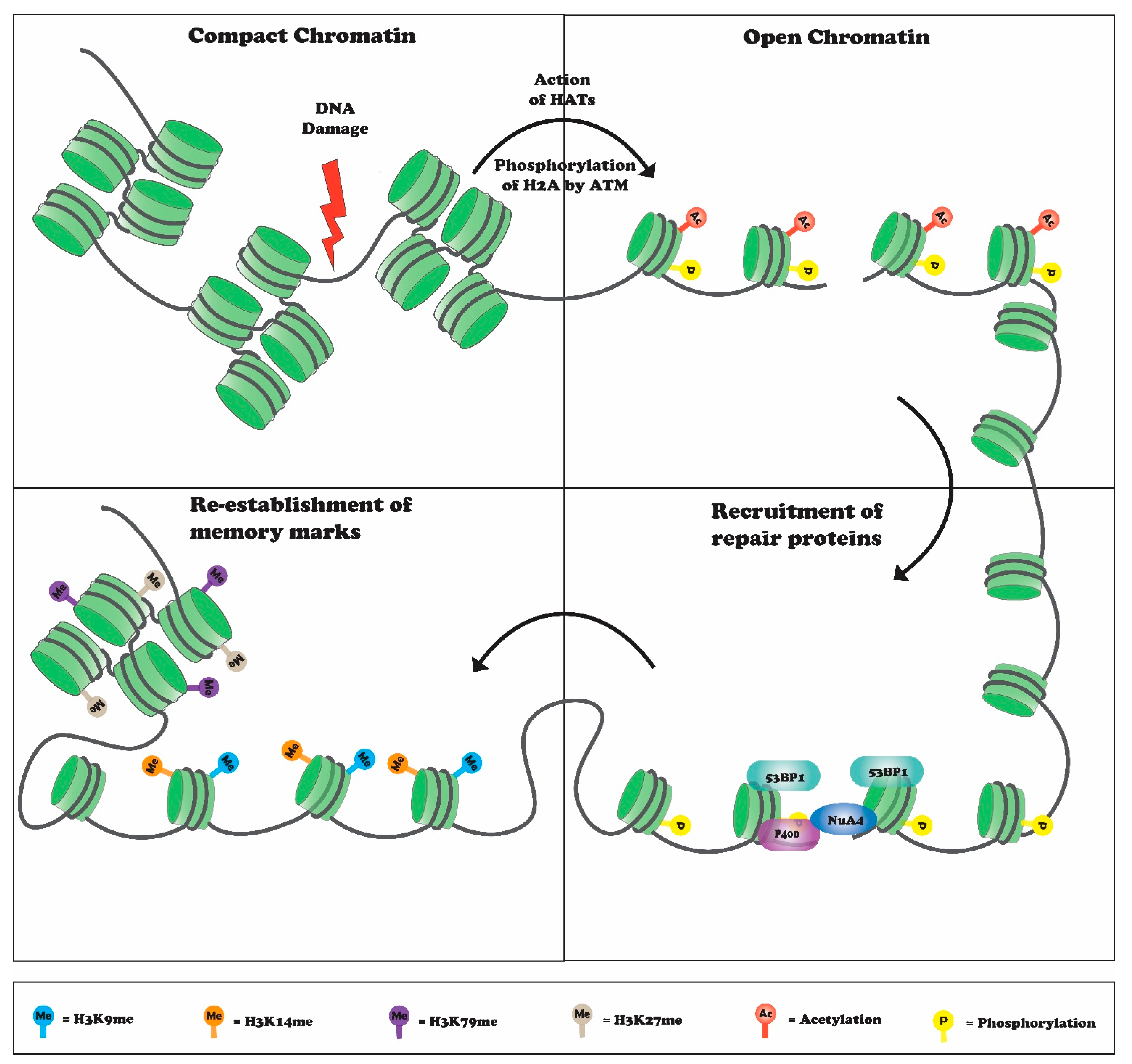
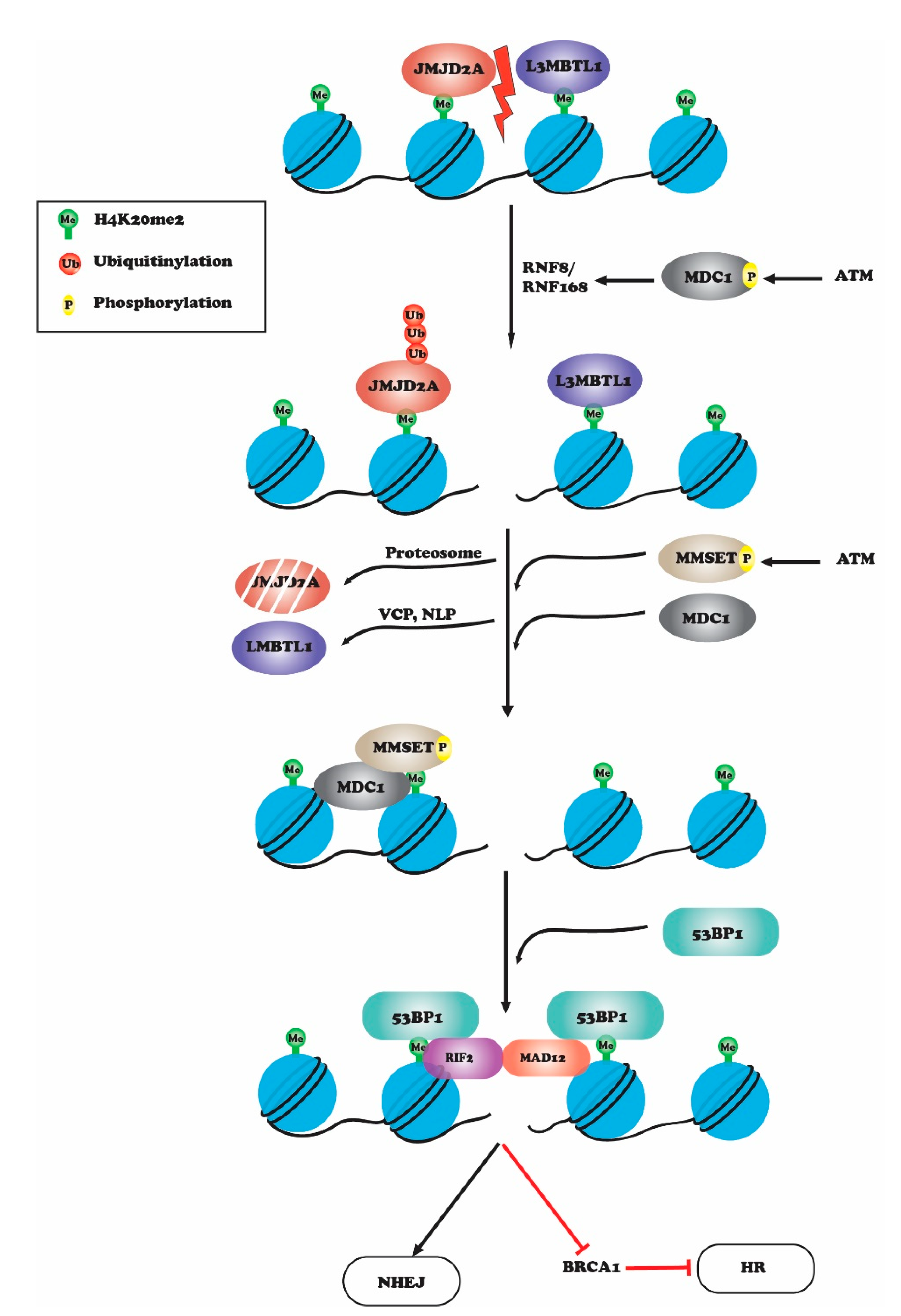
2. Histone H4K20 Methylation in DNA Repair
3. Histone H3K4 Methylation in DNA Repair
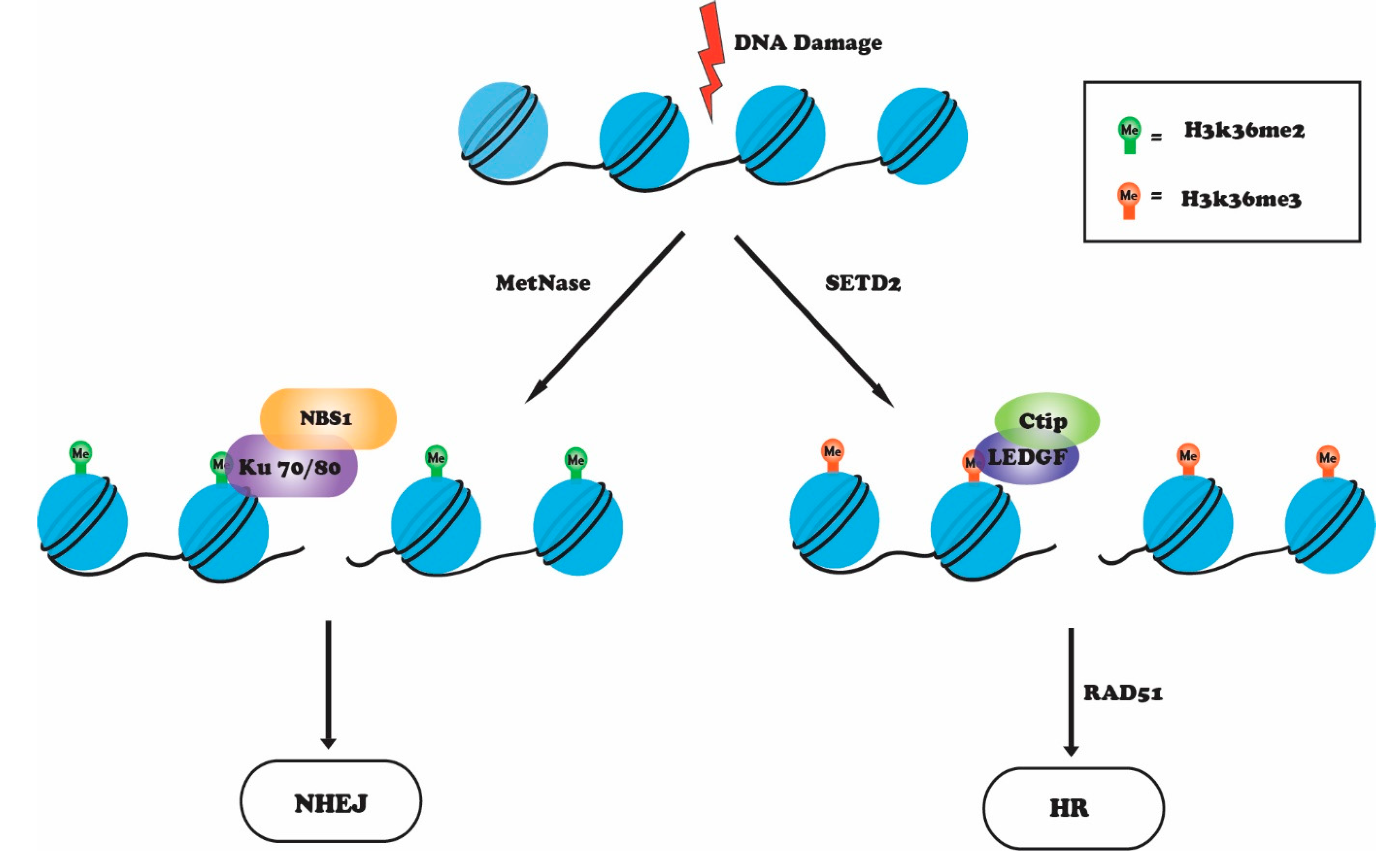
4. H3K36 Methylation in the DNA Damage Response
5. Histone H3K79 Methylation in DNA Repair
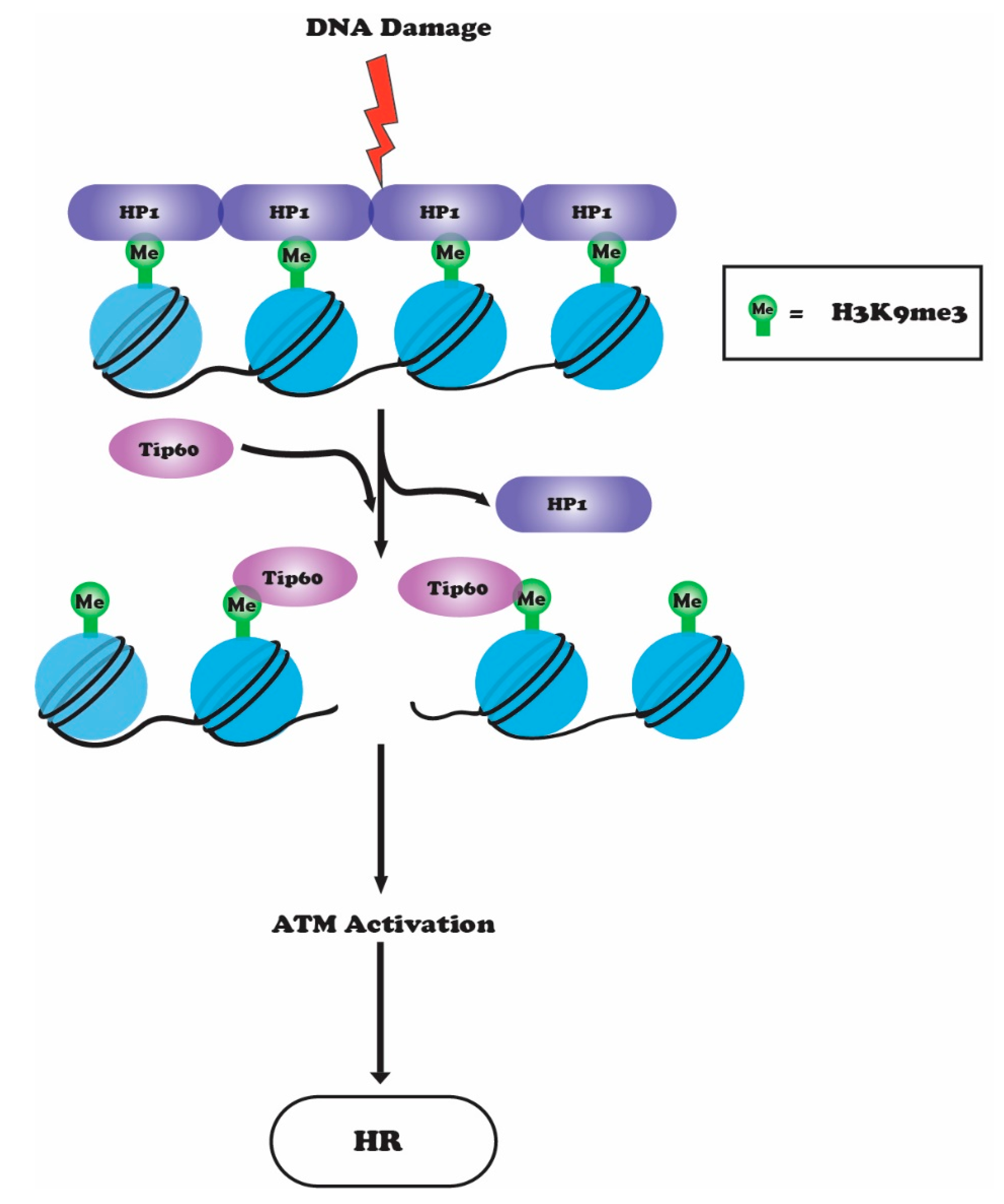
6. Crosstalk between H3K9 Methylation, ATM and TIP60
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farooq, Z.; Banday, S.; Pandita, T.K.; Altaf, M. The many faces of histone H3K79 methylation. Mutat. Res. Rev. Mutat. Res. 2016, 768, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Altaf, M.; Saksouk, N.; Cote, J. Histone modifications in response to DNA damage. Mutat. Res. 2007, 618, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Smeenk, G.; van Attikum, H. The chromatin response to DNA breaks: Leaving a mark on genome integrity. Annu. Rev. BioChem. 2013, 82, 55–80. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Ganai, S.A.; Rashid, R.; Abdullah, E.; Altaf, M. Plant Derived Inhibitor Sulforaphane in Combinatorial Therapy Against Therapeutically Challenging Pancreatic Cancer. Anticancer Agents Med. Chem. 2017, 17, 365–373. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- van Attikum, H.; Gasser, S.M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009, 19, 207–217. [Google Scholar] [CrossRef]
- Helleday, T.; Lo, J.; van Gent, D.C.; Engelward, B.P. DNA double-strand break repair: From mechanistic understanding to cancer treatment. DNA Repair 2007, 6, 923–935. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Haber, J.E. Partners and pathwaysrepairing a double-strand break. Trends Genet. 2000, 16, 259–264. [Google Scholar] [CrossRef]
- Kanaar, R.; Hoeijmakers, J.H.; van Gent, D.C. Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 1998, 8, 483–489. [Google Scholar] [CrossRef]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat. Res. 2017, 803-805, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Van, H.T.; Santos, M.A. Histone modifications and the DNA double-strand break response. Cell Cycle 2018, 17, 2399–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat. Res. 2019, 780, 37–47. [Google Scholar] [CrossRef]
- Pei, H.; Zhang, L.; Luo, K.; Qin, Y.; Chesi, M.; Fei, F.; Bergsagel, P.L.; Wang, L.; You, Z.; Lou, Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 2011, 470, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botuyan, M.V.; Lee, J.; Ward, I.M.; Kim, J.E.; Thompson, J.R.; Chen, J.; Mer, G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 2006, 127, 1361–1373. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.L.; Portoso, M.; Mata, J.; Bahler, J.; Allshire, R.C.; Kouzarides, T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 2004, 119, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Chou, R.H.; Wang, Y.N.; Hsieh, Y.H.; Li, L.Y.; Xia, W.; Chang, W.C.; Chang, L.C.; Cheng, C.C.; Lai, C.C.; Hsu, J.L.; et al. EGFR modulates DNA synthesis and repair through Tyr phosphorylation of histone H4. Dev. Cell 2014, 30, 224–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; de Lange, T. 53BP1: Pro choice in DNA repair. Trends Cell Biol. 2014, 24, 108–117. [Google Scholar] [CrossRef] [Green Version]
- Huyen, Y.; Zgheib, O.; Ditullio, R.A., Jr.; Gorgoulis, V.G.; Zacharatos, P.; Petty, T.J.; Sheston, E.A.; Mellert, H.S.; Stavridi, E.S.; Halazonetis, T.D. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004, 432, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Du, L.L.; Nakamura, T.M.; Russell, P. Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes Dev. 2006, 20, 1583–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trojer, P.; Zhang, J.; Yonezawa, M.; Schmidt, A.; Zheng, H.; Jenuwein, T.; Reinberg, D. Dynamic Histone H1 Isotype 4 Methylation and Demethylation by Histone Lysine Methyltransferase G9a/KMT1C and the Jumonji Domain-containing JMJD2/KDM4 Proteins. J. Biol. Chem. 2009, 284, 8395–8405. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Thompson, J.R.; Botuyan, M.V.; Mer, G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat. Struct Mol. Biol. 2008, 15, 109–111. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Fischle, W.; Wang, W.; Duncan, E.M.; Liang, L.; Murakami-Ishibe, S.; Allis, C.D.; Patel, D.J. Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Mol. Cell 2007, 28, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; Allali-Hassani, A.; Nady, N.; Qi, C.; Ouyang, H.; Liu, Y.; MacKenzie, F.; Vedadi, M.; Arrowsmith, C.H. L3MBTL1 recognition of mono- and dimethylated histones. Nat. Struct Mol. Biol. 2007, 14, 1229–1230. [Google Scholar] [CrossRef]
- Kim, J.; Daniel, J.; Espejo, A.; Lake, A.; Krishna, M.; Xia, L.; Zhang, Y.; Bedford, M.T. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006, 7, 397–403. [Google Scholar] [CrossRef]
- Hsiao, K.Y.; Mizzen, C.A. Histone H4 deacetylation facilitates 53BP1 DNA damage signaling and double-strand break repair. J. Mol. Cell Biol. 2013, 5, 157–165. [Google Scholar] [CrossRef]
- Jacquet, K.; Fradet-Turcotte, A.; Avvakumov, N.; Lambert, J.P.; Roques, C.; Pandita, R.K.; Paquet, E.; Herst, P.; Gingras, A.C.; Pandita, T.K.; et al. The TIP60 Complex Regulates Bivalent Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Mol. Cell 2016, 62, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Mallette, F.A.; Mattiroli, F.; Cui, G.; Young, L.C.; Hendzel, M.J.; Mer, G.; Sixma, T.K.; Richard, S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012, 31, 1865–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acs, K.; Luijsterburg, M.S.; Ackermann, L.; Salomons, F.A.; Hoppe, T.; Dantuma, N.P. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 2011, 18, 1345–1350. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, H.; Chen, Y.; Yang, X.; Wang, P.; Liu, T.; Deng, M.; Qin, B.; Correia, C.; Lee, S.; et al. A cell cycle-dependent BRCA1-UHRF1 cascade regulates DNA double-strand break repair pathway choice. Nat. Commun. 2016, 7, 10201. [Google Scholar] [CrossRef] [PubMed]
- Boersma, V.; Moatti, N.; Segura-Bayona, S.; Peuscher, M.H.; van der Torre, J.; Wevers, B.A.; Orthwein, A.; Durocher, D.; Jacobs, J.J.L. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5’ end resection. Nature 2015, 521, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Sung, P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell Biol. 2014, 34, 1380–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Cho, N.W.; Cui, G.; Manion, E.M.; Shanbhag, N.M.; Botuyan, M.V.; Mer, G.; Greenberg, R.A. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013, 20, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Diaz, C.; Orthwein, A.; Leung, C.C.; Huang, H.; Landry, M.C.; Kitevski-LeBlanc, J.; Noordermeer, S.M.; Sicheri, F.; et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef] [Green Version]
- Shoaib, M.; Walter, D.; Gillespie, P.J.; Izard, F.; Fahrenkrog, B.; Lleres, D.; Lerdrup, M.; Johansen, J.V.; Hansen, K.; Julien, E.; et al. Histone H4K20 methylation mediated chromatin compaction threshold ensures genome integrity by limiting DNA replication licensing. Nat. Commun. 2018, 9, 3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, S.; Schotta, G.; Sorensen, C.S. Histone H4 lysine 20 methylation: Key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013, 41, 2797–2806. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Okamoto, I.; Murphy, N.; Chu, J.; Price, S.M.; Shen, M.M.; Torres-Padilla, M.E.; Heard, E.; Reinberg, D. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol. Cell Biol. 2009, 29, 2278–2295. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, S.; Elvers, I.; Trelle, M.B.; Menzel, T.; Eskildsen, M.; Jensen, O.N.; Helleday, T.; Helin, K.; Sorensen, C.S. The histone methyltransferase SET8 is required for S-phase progression. J. Cell Biol. 2007, 179, 1337–1345. [Google Scholar] [CrossRef] [Green Version]
- Tardat, M.; Murr, R.; Herceg, Z.; Sardet, C.; Julien, E. PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J. Cell Biol. 2007, 179, 1413–1426. [Google Scholar] [CrossRef]
- Mechali, M.; Yoshida, K.; Coulombe, P.; Pasero, P. Genetic and epigenetic determinants of DNA replication origins, position and activation. Curr. Opin. Genet. Dev. 2013, 23, 124–131. [Google Scholar] [CrossRef]
- Prioleau, M.N.; MacAlpine, D.M. DNA replication origins-where do we begin? Genes Dev. 2016, 30, 1683–1697. [Google Scholar] [CrossRef] [Green Version]
- Shikata, D.; Yamamoto, T.; Honda, S.; Ikeda, S.; Minami, N. H4K20 monomethylation inhibition causes loss of genomic integrity in mouse preimplantation embryos. J. Reprod. Dev. 2020, 66, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.L.; Griesenbeck, J.; Kornberg, R.D.; Cleary, M.L. A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc. Natl. Acad. Sci. USA 2002, 99, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Roguev, A.; Schaft, D.; Shevchenko, A.; Pijnappel, W.W.; Wilm, M.; Aasland, R.; Stewart, A.F. The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 2001, 20, 7137–7148. [Google Scholar] [CrossRef]
- Miller, T.; Krogan, N.J.; Dover, J.; Erdjument-Bromage, H.; Tempst, P.; Johnston, M.; Greenblatt, J.F.; Shilatifard, A. COMPASS: A complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. USA 2001, 98, 12902–12907. [Google Scholar] [CrossRef] [Green Version]
- Freitag, M. Histone Methylation by SET Domain Proteins in Fungi. Annu. Rev. Microbiol. 2017, 71, 413–439. [Google Scholar] [CrossRef]
- Faucher, D.; Wellinger, R.J. Methylated H3K4, a transcription-associated histone modification, is involved in the DNA damage response pathway. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, E.Y.; Hong, S.J.; Oum, J.H.; Yanez, Y.; Zhang, Y.; Lee, S.E. RSC mobilizes nucleosomes to improve accessibility of repair machinery to the damaged chromatin. Mol. Cell Biol. 2007, 27, 1602–1613. [Google Scholar] [CrossRef] [Green Version]
- Shim, E.Y.; Ma, J.L.; Oum, J.H.; Yanez, Y.; Lee, S.E. The yeast chromatin remodeler RSC complex facilitates end joining repair of DNA double-strand breaks. Mol. Cell Biol. 2005, 25, 3934–3944. [Google Scholar] [CrossRef] [Green Version]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Rosa, H.; Schneider, R.; Bernstein, B.E.; Karabetsou, N.; Morillon, A.; Weise, C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Methylation of histone H3 K4 mediates association of the Isw1p ATPase with chromatin. Mol. Cell 2003, 12, 1325–1332. [Google Scholar] [CrossRef]
- Nakamura, K.; Kato, A.; Kobayashi, J.; Yanagihara, H.; Sakamoto, S.; Oliveira, D.V.; Shimada, M.; Tauchi, H.; Suzuki, H.; Tashiro, S.; et al. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol. Cell 2011, 41, 515–528. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, W.G. Biological function and regulation of histone and non-histone lysine methylation in response to DNA damage. Acta Biochim. Biophys. Sin. 2016, 48, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Guerillon, C.; Larrieu, D.; Pedeux, R. ING1 and ING2: Multifaceted tumor suppressor genes. Cell Mol. Life Sci. 2013, 70, 3753–3772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wang, L.; Song, P.; Geng, X.; Liang, X.; Zhou, M.; Wang, Y.; Chen, C.; Jia, J.; Zeng, J. Critical role of histone demethylase RBP2 in human gastric cancer angiogenesis. Mol. Cancer 2014, 13, 81. [Google Scholar] [CrossRef] [Green Version]
- Mosammaparast, N.; Kim, H.; Laurent, B.; Zhao, Y.; Lim, H.J.; Majid, M.C.; Dango, S.; Luo, Y.; Hempel, K.; Sowa, M.E.; et al. The histone demethylase LSD1/KDM1A promotes the DNA damage response. J. Cell Biol. 2013, 203, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, L.; Yang, S.; Song, N.; Zhou, X.; Gao, J.; Yu, N.; Shan, L.; Wang, Q.; Liang, J.; et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc. Natl. Acad. Sci. USA 2014, 111, 7096–7101. [Google Scholar] [CrossRef] [Green Version]
- Gong, F.; Clouaire, T.; Aguirrebengoa, M.; Legube, G.; Miller, K.M. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J. Cell Biol. 2017, 216, 1959–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriks, I.A.; Vertegaal, A.C. SUMO in the DNA damage response. Oncotarget 2015, 6, 15734–15735. [Google Scholar] [CrossRef]
- DiFiore, J.V.; Ptacek, T.S.; Wang, Y.; Li, B.; Simon, J.M.; Strahl, B.D. Unique and Shared Roles for Histone H3K36 Methylation States in Transcription Regulation Functions. Cell Rep. 2020, 31, 107751. [Google Scholar] [CrossRef]
- Kizer, K.O.; Phatnani, H.P.; Shibata, Y.; Hall, H.; Greenleaf, A.L.; Strahl, B.D. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol. Cell Biol. 2005, 25, 3305–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.M. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, S.; Smolle, M.; Li, H.; Gogol, M.M.; Saint, M.; Kumar, S.; Natarajan, K.; Workman, J.L. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature 2012, 489, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Carrozza, M.J.; Li, B.; Florens, L.; Suganuma, T.; Swanson, S.K.; Lee, K.K.; Shia, W.J.; Anderson, S.; Yates, J.; Washburn, M.P.; et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 2005, 123, 581–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, D.K.; Strahl, B.D. An RNA polymerase II-coupled function for histone H3K36 methylation in checkpoint activation and DSB repair. Nat. Commun. 2014, 5, 3965. [Google Scholar] [CrossRef] [Green Version]
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, S.X.; Ahrabi, S.; Zalmas, L.P.; Sarkar, S.; Aymard, F.; Bachrati, C.Z.; Helleday, T.; Legube, G.; La Thangue, N.B.; Porter, A.C.; et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014, 7, 2006–2018. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, S.; Vitor, A.C.; Sridhara, S.C.; Martins, F.B.; Raposo, A.C.; Desterro, J.M.; Ferreira, J.; de Almeida, S.F. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife 2014, 3, e02482. [Google Scholar] [CrossRef]
- Daugaard, M.; Baude, A.; Fugger, K.; Povlsen, L.K.; Beck, H.; Sorensen, C.S.; Petersen, N.H.; Sorensen, P.H.; Lukas, C.; Bartek, J.; et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 2012, 19, 803–810. [Google Scholar] [CrossRef]
- Bannister, A.J.; Schneider, R.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J. Biol. Chem. 2005, 280, 17732–17736. [Google Scholar] [CrossRef] [Green Version]
- Fnu, S.; Williamson, E.A.; De Haro, L.P.; Brenneman, M.; Wray, J.; Shaheen, M.; Radhakrishnan, K.; Lee, S.H.; Nickoloff, J.A.; Hromas, R. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl. Acad. Sci. USA 2011, 108, 540–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strahl, B.D.; Grant, P.A.; Briggs, S.D.; Sun, Z.W.; Bone, J.R.; Caldwell, J.A.; Mollah, S.; Cook, R.G.; Shabanowitz, J.; Hunt, D.F.; et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell Biol. 2002, 22, 1298–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.; Li, C.; Yin, Z.; Wen, J.; Meng, H.; Xue, L.; Wang, J. Histone methylation in DNA repair and clinical practice: New findings during the past 5-years. J. Cancer 2018, 9, 2072–2081. [Google Scholar] [CrossRef]
- Hromas, R.; Williamson, E.A.; Fnu, S.; Lee, Y.J.; Park, S.J.; Beck, B.D.; You, J.S.; Leitao, A.; Nickoloff, J.A.; Lee, S.H. Chk1 phosphorylation of Metnase enhances DNA repair but inhibits replication fork restart. Oncogene 2012, 31, 4245–4254. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.F.; Chu, P.C.; Wu, P.Y.; Yu, M.Y.; Lee, J.Y.; Tsai, M.D.; Chang, M.S. PHRF1 promotes genome integrity by modulating non-homologous end-joining. Cell Death Dis. 2015, 6, e1716. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.H.; Feng, Q.; Wang, H.; Erdjument-Bromage, H.; Tempst, P.; Zhang, Y.; Struhl, K. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002, 16, 1518–1527. [Google Scholar] [CrossRef] [Green Version]
- Altaf, M.; Utley, R.T.; Lacoste, N.; Tan, S.; Briggs, S.D.; Cote, J. Interplay of chromatin modifiers on a short basic patch of histone H4 tail defines the boundary of telomeric heterochromatin. Mol. Cell 2007, 28, 1002–1014. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Chiariello, N.R.; Tobeck, T.; Fukami, T.; Field, C.B. Nonlinear, interacting responses to climate limit grassland production under global change. Proc. Natl. Acad. Sci. USA 2016, 113, 10589–10594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FitzGerald, J.; Moureau, S.; Drogaris, P.; O’Connell, E.; Abshiru, N.; Verreault, A.; Thibault, P.; Grenon, M.; Lowndes, N.F. Regulation of the DNA damage response and gene expression by the Dot1L histone methyltransferase and the 53Bp1 tumour suppressor. PLoS ONE 2011, 6, e14714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysocki, R.; Javaheri, A.; Allard, S.; Sha, F.; Cote, J.; Kron, S.J. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol. Cell Biol. 2005, 25, 8430–8443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usui, T.; Shinohara, A. Rad9, a 53BP1 Ortholog of Budding Yeast, Is Insensitive to Spo11-Induced Double-Strand Breaks during Meiosis. Front. Cell Dev. Biol. 2021, 9, 635383. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [Green Version]
- Wakeman, T.P.; Wang, Q.; Feng, J.; Wang, X.F. Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA damage-induced 53BP1 foci at G1/G2 cell-cycle phases. EMBO J. 2012, 31, 2169–2181. [Google Scholar] [CrossRef] [Green Version]
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Conde, F.; Refolio, E.; Cordon-Preciado, V.; Cortes-Ledesma, F.; Aragon, L.; Aguilera, A.; San-Segundo, P.A. The Dot1 histone methyltransferase and the Rad9 checkpoint adaptor contribute to cohesin-dependent double-strand break repair by sister chromatid recombination in Saccharomyces cerevisiae. Genetics 2009, 182, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Chen, S.; Wang, H.; Yin, C.; Han, C.; Peng, C.; Liu, Z.; Wan, L.; Zhang, X.; Zhang, J.; et al. The protective role of DOT1L in UV-induced melanomagenesis. Nat. Commun. 2018, 9, 259. [Google Scholar] [CrossRef]
- Mandemaker, I.K.; Vermeulen, W.; Marteijn, J.A. Gearing up chromatin: A role for chromatin remodeling during the transcriptional restart upon DNA damage. Nucleus 2014, 5, 203–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Alagoz, M.; Katsuki, Y.; Ogiwara, H.; Ogi, T.; Shibata, A.; Kakarougkas, A.; Jeggo, P. SETDB1, HP1 and SUV39 promote repositioning of 53BP1 to extend resection during homologous recombination in G2 cells. Nucleic Acids Res. 2015, 43, 7931–7944. [Google Scholar] [CrossRef] [Green Version]
- Ayrapetov, M.K.; Gursoy-Yuzugullu, O.; Xu, C.; Xu, Y.; Price, B.D. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. USA 2014, 111, 9169–9174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Jiang, X.; Xu, Y.; Ayrapetov, M.K.; Moreau, L.A.; Whetstine, J.R.; Price, B.D. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat. Cell Biol. 2009, 11, 1376–1382. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.C.; Karpen, G.H. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat. Cell Biol. 2007, 9, 25–35. [Google Scholar] [CrossRef]
- Mozzetta, C.; Boyarchuk, E.; Pontis, J.; Ait-Si-Ali, S. Sound of silence: The properties and functions of repressive Lys methyltransferases. Nat. Rev. Mol. Cell Biol. 2015, 16, 499–513. [Google Scholar] [CrossRef]
- Padeken, J.; Zeller, P.; Towbin, B.; Katic, I.; Kalck, V.; Methot, S.P.; Gasser, S.M. Synergistic lethality between BRCA1 and H3K9me2 loss reflects satellite derepression. Genes Dev. 2019, 33, 436–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Hoong, N.; Aslanian, A.; Hara, T.; Benner, C.; Heinz, S.; Miga, K.H.; Ke, E.; Verma, S.; Soroczynski, J.; et al. Heterochromatin-Encoded Satellite RNAs Induce Breast Cancer. Mol. Cell 2018, 70, 842–853.e7. [Google Scholar] [CrossRef] [Green Version]
- Zeller, P.; Padeken, J.; van Schendel, R.; Kalck, V.; Tijsterman, M.; Gasser, S.M. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat. Genet. 2016, 48, 1385–1395. [Google Scholar] [CrossRef]
- Fortuny, A.; Chansard, A.; Caron, P.; Chevallier, O.; Leroy, O.; Renaud, O.; Polo, S.E. Imaging the response to DNA damage in heterochromatin domains reveals core principles of heterochromatin maintenance. Nat. Commun. 2021, 12, 2428. [Google Scholar] [CrossRef]
- Khoury-Haddad, H.; Guttmann-Raviv, N.; Ipenberg, I.; Huggins, D.; Jeyasekharan, A.D.; Ayoub, N. PARP1-dependent recruitment of KDM4D histone demethylase to DNA damage sites promotes double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, E728–E737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.C.; McDonald, D.W.; Hendzel, M.J. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following gamma-irradiation. J. Biol. Chem. 2013, 288, 21376–21388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, H.Y.; Hussain, A.; Qi, J. Role of H3K9 demethylases in DNA double-strand break repair. J. Cancer Biol. 2020, 1, 10–15. [Google Scholar] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mushtaq, A.; Mir, U.S.; Hunt, C.R.; Pandita, S.; Tantray, W.W.; Bhat, A.; Pandita, R.K.; Altaf, M.; Pandita, T.K. Role of Histone Methylation in Maintenance of Genome Integrity. Genes 2021, 12, 1000. https://doi.org/10.3390/genes12071000
Mushtaq A, Mir US, Hunt CR, Pandita S, Tantray WW, Bhat A, Pandita RK, Altaf M, Pandita TK. Role of Histone Methylation in Maintenance of Genome Integrity. Genes. 2021; 12(7):1000. https://doi.org/10.3390/genes12071000
Chicago/Turabian StyleMushtaq, Arjamand, Ulfat Syed Mir, Clayton R. Hunt, Shruti Pandita, Wajahat W. Tantray, Audesh Bhat, Raj K. Pandita, Mohammad Altaf, and Tej K. Pandita. 2021. "Role of Histone Methylation in Maintenance of Genome Integrity" Genes 12, no. 7: 1000. https://doi.org/10.3390/genes12071000