
Genomic Signatures of Domestication Selection in the Australasian Snapper (Chrysophrys auratus)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Populations: Pedigree Structure and Information
2.2. Sampling and Generation of Molecular Data
2.3. Raw Reads, Mapping, Variant Calling, and Filtering of the Data
2.4. Analysis of Genetic Differentiation between Generations
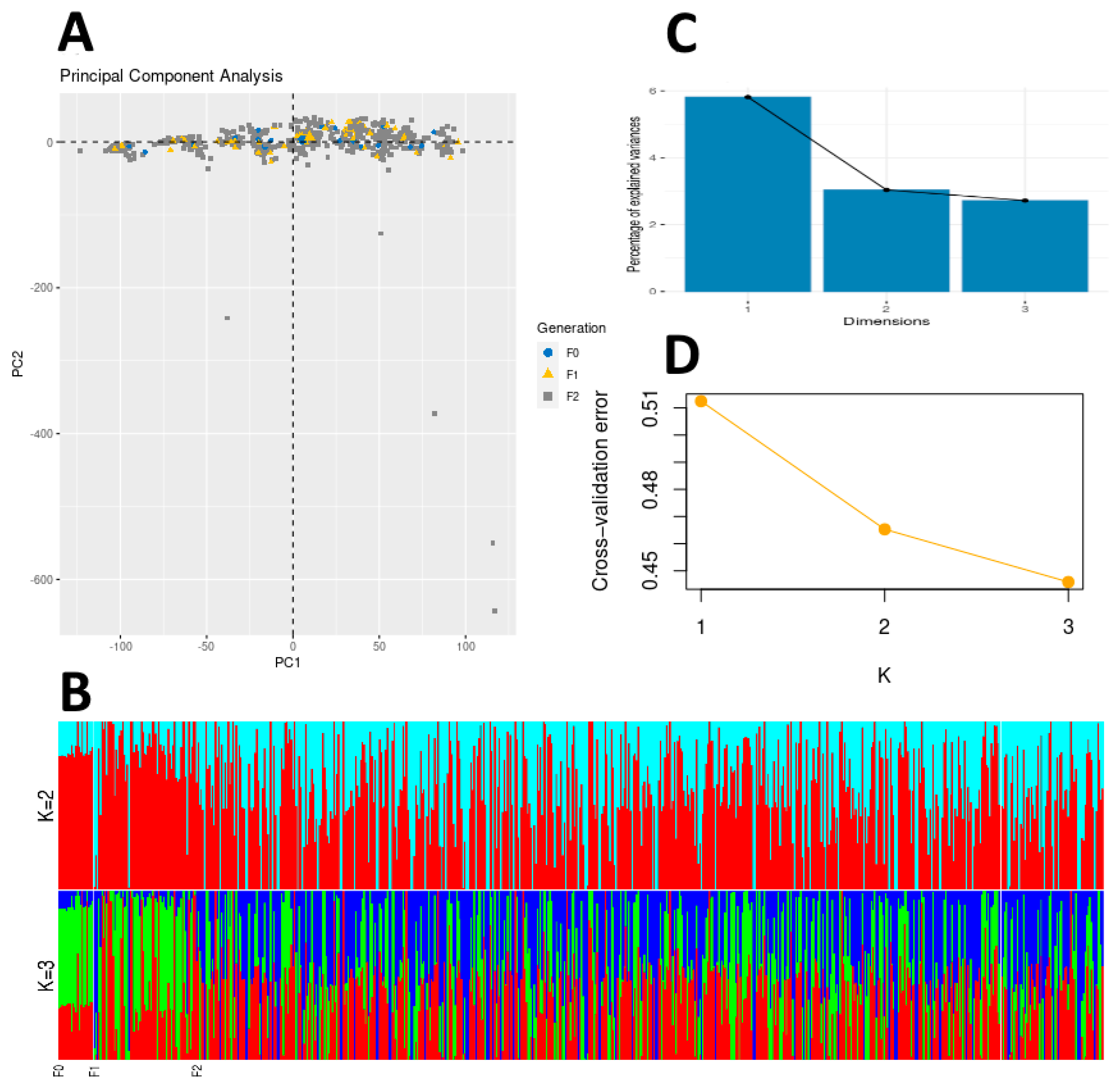
2.5. ADMIXTURE Analysis
2.6. Principal Component Analysis
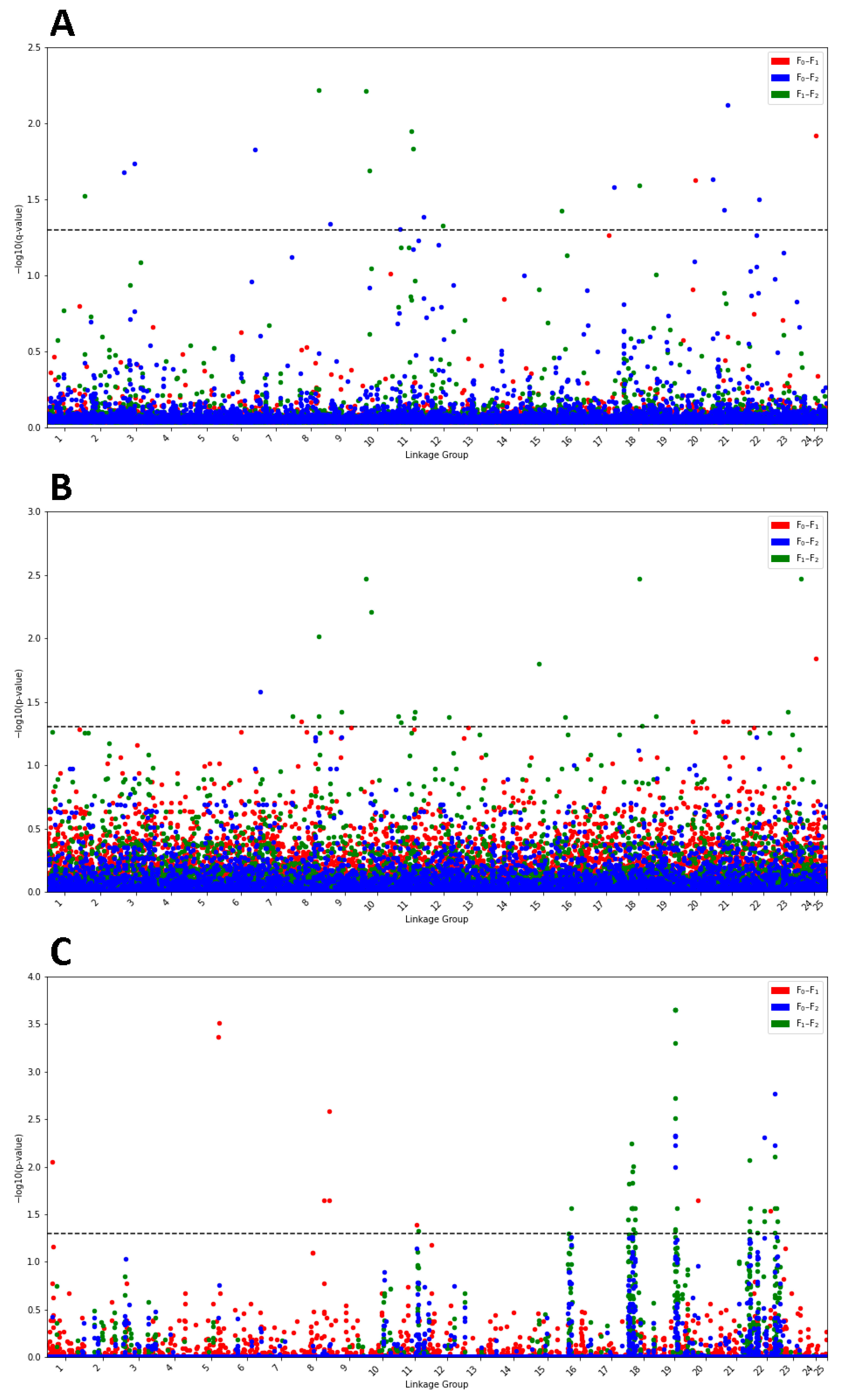
2.7. Identifying SNP Variants Associated with Selective Sweeps
2.8. Annotation of Genes near Putative Variants
3. Results
3.1. Genotyping and Quality Control
3.2. Analysis of Genetic Differentiation between Generations
3.3. ADMIXTURE Analysis
3.4. Principal Component Analysis
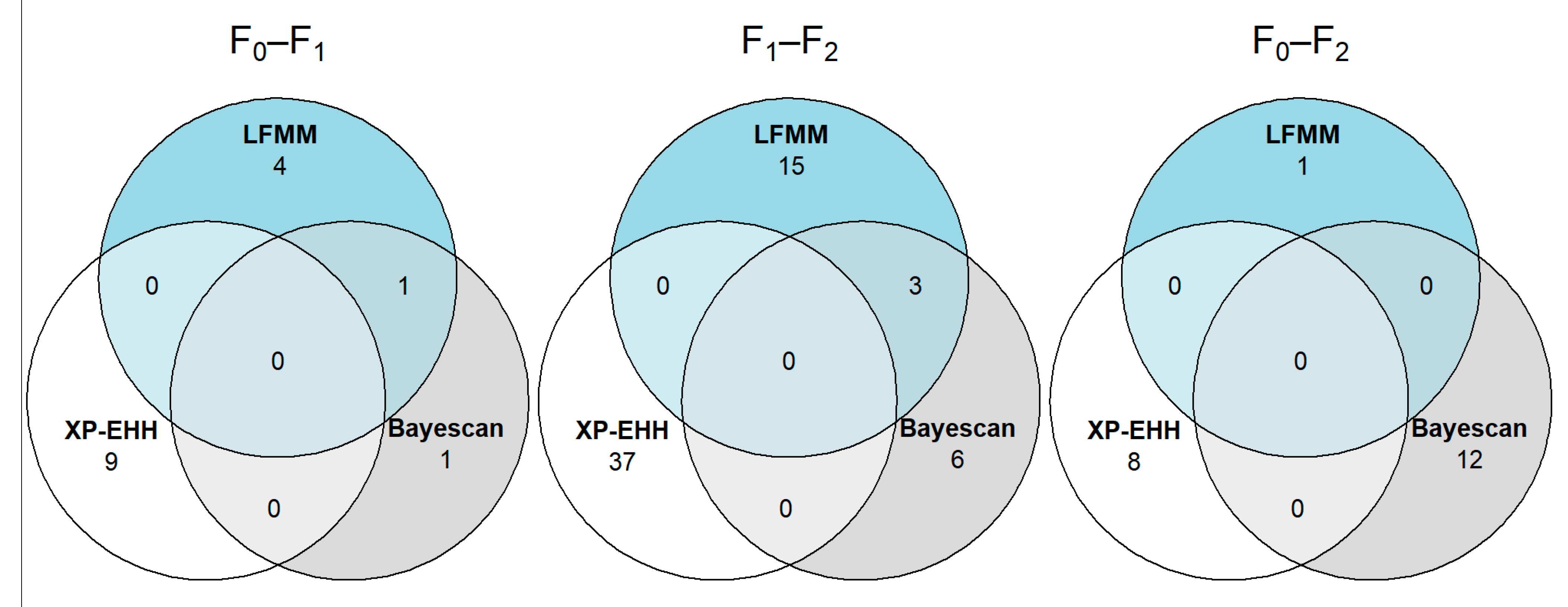
3.5. Identifying SNP Variants Associated with Selective Sweeps
3.6. Annotation of Genes near Putative Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zeder, M.A. Core questions in domestication research. Proc. Natl. Acad. Sci. USA 2015, 112, 3191–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchings, J.A.; Fraser, D.J. The nature of fisheries-and farming-induced evolution. Mol. Ecol. 2008, 17, 294–313. [Google Scholar] [CrossRef]
- Gjedrem, T.; Robinson, N. Advances by Selective Breeding for Aquatic Species: A Review. Agric. Sci. 2014, 5, 1152–1158. [Google Scholar] [CrossRef] [Green Version]
- Gjedrem, T.; Robinson, N.; Rye, M. The importance of selective breeding in aquaculture to meet future demands for animal protein: A review. Aquaculture 2012, 350–353, 117–129. [Google Scholar] [CrossRef]
- Stephan, W. Signatures of positive selection: From selective sweeps at individual loci to subtle allele frequency changes in polygenic adaptation. Mol. Ecol. 2015, 25, 79–88. [Google Scholar] [CrossRef]
- Pritchard, J.K.; di Rienzo, A. Adaptation—Not by sweeps alone. Nat. Rev. Genet. 2010, 11, 665–667. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Admetlla, A.; Liang, M.; Korneliussen, T.S.; Nielsen, R. On Detecting Incomplete Soft or Hard Selective Sweeps Using Haplotype Structure. Mol. Biol. Evol. 2014, 31, 1275–1291. [Google Scholar] [CrossRef] [Green Version]
- Ellegren, H. Genome sequencing and population genomics in non-model organisms. Trends Ecol. Evol. 2014, 29, 51–63. [Google Scholar] [CrossRef]
- Haasl, R.J.; Payseur, B.A. Fifteen years of genomewide scans for selection: Trends, lessons and unaddressed genetic sources of complication. Mol. Ecol. 2016, 25, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Symonds, J.E.; King, N.; Camara, M.D.; Ragg, N.L.C.; Hilton, Z.; Walker, S.P.; Roberts, R.; Malpot, E.; Preece, M.; Amer, P.R.; et al. New Zealand aquaculture selective breeding: From theory to industry application for three flagship species. In Proceedings of the World Congress on Genetics Applied to Livestock Production, Lincoln, NE, USA, 16–22 July 1986. [Google Scholar]
- Wellenreuther, M.; Le Luyer, J.; Cook, D.; Ritchie, P.A.; Bernatchez, L. Domestication and Temperature Modulate Gene Expression Signatures and Growth in the Australasian Snapper Chrysophrys auratus. G3 Genes|Genomes|Genetics 2019, 9, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Ashton, D.T.; Ritchie, P.A.; Wellenreuther, M. High-density linkage map and QTLs for growth in snapper (Chrysophrys auratus). G3 Genes Genomes Genet. 2019, 9, 1027–1035. [Google Scholar] [CrossRef] [Green Version]
- Ashton, D.T.; Hilario, P.; Jaksons, P.A.; Wellenreuther, R.M. Genetic diversity and heritability of economically important traits in the Australasian snapper (Chrysophrys auratus). Aquaculture 2019, 505, 190–198. [Google Scholar] [CrossRef]
- Smith, P.J.; Francis, R.I.C.C.; Paul, L.J. Genetic variation and population structure in the New Zealand snapper. N. Z. J. Mar. Freshw. Res. 1978, 12, 343–350. [Google Scholar] [CrossRef]
- Catanach, A.; Crowhurst, R.; Deng, C.; David, C.; Bernatchez, L.; Wellenreuther, M. The genomic pool of standing structural variation outnumbers single nucleotide polymorphism by threefold in the marine teleost Chrysophrys auratus. Mol. Ecol. 2019, 28, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Knutsen, H.; Olsen, E.M.; Jorde, P.E.; Espeland, S.H.; Andre, C.; Stenseth, N.C. Are low but statistically significant levels of genetic differentiation in marine fishes ‘biologically meaningful’? A case study of coastal Atlantic cod. Mol. Ecol. 2010, 20, 768–783. [Google Scholar] [CrossRef] [PubMed]
- McKeown, N.J.; Arkhipkin, A.I.; Shaw, P.W. Regional genetic population structure and fine scale genetic cohesion in the Southern blue whiting Micromesistius australis. Fish. Res. 2017, 185, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, H.B.; Hansen, M.M.; Bekkevold, D.; Ruzzante, D.E.; Loeschcke, V. Marine landscapes and population genetic structure of herring (Clupea harengus L.) in the Baltic Sea. Mol. Ecol. 2005, 14, 3219–3234. [Google Scholar] [CrossRef]
- Sandoval, J.; Beheregaray, L.; Wellenreuther, M. Genomic prediction of growth in a commercially, recreationally, and culturally important marine resource, the Australian snapper (Chrysophrys auratus). G3 Genes Genomes Genet. 2021, jkab361. [Google Scholar] [CrossRef]
- Irving, K.; Wellenreuther, M.; Ritchie, P.A. Description of the growth hormone gene of the Australasian snapper, Chrysophrys auratus, and associated intra-and interspecific genetic variation. J. Fish Biol. 2021, 99, 1060–1070. [Google Scholar] [CrossRef]
- Elshire, R.; Glaubitz, J.C.; Sun, Q.; Poland, J.; Kawamoto, K.; Buckler, E.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: Building and genotyping loci de novo from short-read sequences. G3 Genes Genomes Genet. 2011, 1, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- O’Leary, S.J.; Puritz, J.B.; Willis, S.C.; Hollenbeck, C.M.; Portnoy, D.S. These Aren’t the Loci You’re Looking for: Principles of Effective SNP Filtering for Molecular Ecologists; Wiley Online Library: Hoboken, NJ, USA, 2018. [Google Scholar]
- Roesti, M.; Salzburger, W.; Berner, D. Uninformative polymorphisms bias genome scans for signatures of selection. BMC Evol. Biol. 2012, 12, 94. [Google Scholar] [CrossRef] [Green Version]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodöhl, P.A. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New tools for the analysis of genome-wide SNA data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [Green Version]
- Lischer, H.E.L.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2011, 28, 298–299. [Google Scholar] [CrossRef] [Green Version]
- Foll, M.; Gaggiotti, O. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genet 2008, 180, 977–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caye, K.; Jumentier, B.; Lepeule, J.; François, O. LFMM 2: Fast and accurate inference of gene-environment associations in genome-wide studies. Mol. Biol. Evol. 2019, 36, 852–860. [Google Scholar] [CrossRef]
- Frichot, E.; Schoville, S.D.; Bouchard, G.; François, O. Testing for associations between loci and environmental gradients using latent factor mixed models. Mol. Biol. Evol. 2013, 30, 1687–1699. [Google Scholar] [CrossRef] [Green Version]
- Rellstab, C.; Gugerli, F.; Eckert, A.J.; Hancock, A.; Holderegger, R. A practical guide to environmental association analysis in landscape genomics. Mol. Ecol. 2015, 24, 4348–4370. [Google Scholar] [CrossRef] [Green Version]
- Frichot, E.; François, O. LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 2015, 6, 925–929. [Google Scholar] [CrossRef]
- Dabney, A.; Storey, J.D.; Warnes, G. Qvalue: Q-Value Estimation for False Discovery Rate Control; R Package Version; R Foundation for Statistical Computing: Vienna, Austria, 2010; Volume 1. [Google Scholar]
- Gautier, M.; Klassmann, A.; Vitalis, R. Rehh 2.0: A reimplementation of the R package rehh to detect positive selection from haplotype structure. Mol. Ecol. Res. 2017, 17, 78–90. [Google Scholar] [CrossRef]
- Gautier, M.; Vitalis, R. Rehh: An R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 2012, 28, 1176–1177. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M.; Klassmann, A.; Vitalis, R. Package ‘Rehh’; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Larsson, J. 2019 Eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses. R Package Version 6.1.0. CRAN—Package Eulerr. Available online: r-project.org (accessed on 15 November 2020).
- Quinlan, A.R. BEDTools: The Swiss—Army tool for genome feature analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Lopez Dinamarca, M.E.; Neira, R.; Yáñez, J.M. Applications in the search for genomic selection signatures in fish. Front. Genet. 2015, 5, 458. [Google Scholar]
- Ahrens, C.W.; Rymer, P.D.; Stow, A.; Bragg, J.; Dillon, S.; Umbers, K.D.L.; Dudaniec, R.Y. The search for loci under selection: Trends, biases and progress. Mol. Ecol. 2018, 27, 1342–1356. [Google Scholar] [CrossRef] [Green Version]
- Lotterhos, K.E.; Whitlock, M.C. Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Mol. Ecol. 2014, 23, 2178–2192. [Google Scholar] [CrossRef] [Green Version]
- López, M.E.; Benestan, L.; Moore, J.S.; Perrier, C.; Gilbey, J.; di Genova, A.; Maass, A.; Diaz, D.; Lhorente, J.P.; Correa, K. Comparing genomic signatures of domestication in two Atlantic salmon (Salmo salar L.) populations with different geographical origins. Evol. Appl. 2019, 12, 137–156. [Google Scholar] [CrossRef] [Green Version]
- François, O.; Martins, H.; Caye, K.; Schoville, S. Controlling false discoveries in genome scans for selection. Mol. Ecol. 2016, 25, 454–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellenreuther, M.; Hansson, B. Detecting polygenic evolution: Problems, pitfalls, and promises. Trends Genet. 2016, 32, 155–164. [Google Scholar] [CrossRef]
- Gutierrez, A.; Yáñez, J.; Davidson, W. Evidence of recent signatures of selection during domestication in an Atlantic salmon population. Mar. Genom. 2016, 26, 41–50. [Google Scholar] [CrossRef]
- López, M.E.; Linderoth, T.; Norris, A.; Lhorente, J.P.; Neira, R.; Yáñez, J.M. Multiple Selection Signatures in Farmed Atlantic Salmon Adapted to Different Environments Across Hemispheres. Front. Genet. 2019, 10, 901. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.; Zhou, G.; Guo, J.; Yan, H.; Niu, Y.; Li, Y.; Yuan, C.; Geng, R.; Lan, X.; et al. Whole-genome sequencing of eight goat populations for the detection of selection signatures underlying production and adaptive traits. Sci. Rep. 2016, 6, 38932. [Google Scholar] [CrossRef] [Green Version]
- Schleinitz, D.; Klöting, N.; Lindgren, C.; Breitfeld, J.; Dietrich, A.; Schön, M.R.; Lohmann, T.; Dreßler, M.; Stumvoll, M.; McCarthy, M.; et al. Fat depot-specific mRNA expression of novel loci associated with waist–hip ratio. Int. J. Obes. 2013, 38, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Fritzsche, S.; Kenzelmann, M.; Hoffmann, M.J.; Müller, M.; Engers, R.; Gröne, H.-J.; Schulz, W. Concomitant down-regulation of SPRY1 and SPRY2 in prostate carcinoma. Endocr. -Relat. Cancer 2006, 13, 839–849. [Google Scholar] [CrossRef] [Green Version]
- Tong, D.-L.; Chen, R.-G.; Lu, Y.-L.; Li, W.-K.; Zhang, Y.-F.; Lin, J.-K.; He, L.-J.; Dang, T.; Shan, S.-F.; Xu, X.-H.; et al. The critical role of ASD-related gene CNTNAP3 in regulating synaptic development and social behavior in mice. Neurobiol. Dis. 2019, 130, 104486. [Google Scholar] [CrossRef]
- Kukekova, A.V.; Johnson, J.L.; Xiang, X.; Feng, S.; Liu, S.; Rando, H.M.; Kharlamova, A.V.; Herbeck, Y.; Serdyukova, N.A.; Xiong, Z.; et al. Red fox genome assembly identifies genomic regions associated with tame and aggressive behaviours. Nat. Ecol. Evol. 2018, 2, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Putri, R.; Priyanto, R.; Gunawan, A. Association of Calpastatin (CAST) gene with growth traits and carcass characteristics in Bali cattle. Media Peternak. 2015, 38, 145–149. [Google Scholar] [CrossRef]
- Gebreselassie, G.; Berihulay, H.; Jiang, L.; Ma, Y. Review on Genomic Regions and Candidate Genes Associated with Economically Important Production and Reproduction Traits in Sheep (Ovies aries). Animals 2019, 10, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, C.A.; Parkin, C.A.; Bidet, Y.; Ingham, P.W. A role for the Myoblast city homologues Dock1 and Dock5 and the adaptor proteins Crk and Crk-like in zebrafish myoblast fusion. Development 2007, 134, 3145–3153. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, F.C.; Peixoto, M.G.C.D.; Fonseca, P.A.D.S.; Pires, M.D.F.; Ventura, R.V.; Rosse, I.D.C.; Bruneli, F.A.T.; Machado, M.A.; Carvalho, M.R.S. Identification of Candidate Genes for Reactivity in Guzerat (Bos indicus) Cattle: A Genome-Wide Association Study. PLoS ONE 2017, 12, e0169163. [Google Scholar] [CrossRef] [Green Version]
- Boccardo, A.; Marelli, S.P.; Pravettoni, D.; Bagnato, A.; Busca, G.A.; Strillacci, M.G. The German Shorthair Pointer Dog Breed (Canis lupus familiaris): Genomic Inbreeding and Variability. Animals 2020, 10, 498. [Google Scholar] [CrossRef] [Green Version]
- Kim, O.-H.; Cho, H.-J.; Han, E.; Hong, T.I.; Ariyasiri, K.; Choi, J.-H.; Hwang, K.-S.; Jeong, Y.-M.; Yang, S.-Y.; Yu, K.; et al. Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism. Mol. Autism 2017, 8, 50. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, W.; Yang, B.; Zhang, Z.; Ai, H.; Ren, J.; Huang, L. Signatures of Selection and Interspecies Introgression in the Genome of Chinese Domestic Pigs. Genome Biol. Evol. 2017, 9, 2592–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Yao, J.; Liu, S.; Jiang, C.; Zhang, J.; Li, Y.; Feng, J.; Liu, Z. Identification and expression analysis of 26 oncogenes of the receptor tyrosine kinase family in channel catfish after bacterial infection and hypoxic stress. Comp. Biochem. Physiol. Part D Genom. Proteom. 2015, 14, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Ramalho-Oliveira, R.; Oliveira-Vieira, B.; Viola, J.P. IRF2BP2: A new player in the regulation of cell homeostasis. J. Leukoc. Biol. 2019, 106, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-K.; Qu, H.; Gao, D.; Di, W.; Chen, H.-W.; Guo, X.; Zhai, Z.-H.; Chen, D.-Y. ARF-like Protein 16 (ARL16) Inhibits RIG-I by Binding with Its C-terminal Domain in a GTP-dependent Manner. J. Biol. Chem. 2011, 286, 10568–10580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Basang, Z.; Ding, H.; Lu, Z.; Ning, T.; Wei, H.; Cai, H.; Ke, Y. Latexin expression is downregulated in human gastric carcinomas and exhibits tumor suppressor potential. BMC Cancer 2011, 11, 121. [Google Scholar] [CrossRef] [Green Version]
- Marastoni, S.; Andreuzzi, E.; Paulitti, A.; Colladel, R.; Pellicani, R.; Todaro, F.; Schiavinato, A.; Bonaldo, P.; Colombatti, A.; Mongiat, M. EMILIN2 down-modulates the Wnt signalling pathway and suppresses breast cancer cell growth and migration. J. Pathol. 2013, 232, 391–404. [Google Scholar] [CrossRef]
- Su, G.; Hilgers, W.; Shekher, M.C.; Tang, D.J.; Yeo, C.J.; Hruban, R.H.; Kern, S.E. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor suppressor gene. Cancer Res. 1998, 58, 2339–2342. [Google Scholar]
- McCormick, C.; Duncan, G.; Goutsos, K.T.; Tufaro, F. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc. Natl. Acad. Sci. USA 2000, 97, 668–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carapito, R.; Ivanova, E.L.; Morlon, A.; Meng, L.; Molitor, A.; Erdmann, E.; Kieffer, B.; Pichot, A.; Naegely, L.; Kolmer, A.; et al. ZMIZ1 variants cause a syndromic neurodevelopmental disorder. Am. J. Hum. Genet. 2019, 104, 319–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kähler, A.K.; Djurovic, S.; Kulle, B.; Jönsson, E.G.; Agartz, I.; Hall, H.; Opjordsmoen, S.; Jakobsen, K.D.; Hansen, T.; Melle, I.; et al. Association analysis of schizophrenia on 18 genes involved in neuronal migration: MDGA1 as a new susceptibility gene. Am. J. Med Genet. Part B 2008, 147, 1089–1100. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| A | B | |||||
|---|---|---|---|---|---|---|
| N | HE | HO | FIS (95% CI) | FST (95% CI) | ||
| F0 | 22 | 0.31 | 0.31 | −0.024 (0.04/0.012) | F0–F1 | 0.0085 (0.0021/0.0211) |
| F1 | 65 | 0.32 | 0.33 | −0.049 (0.062/0.035) | F1–F2 | 0.0214 (0.0201/0.0231) |
| F2 | 575 | 0.32 | 0.34 | −0.064 (0.071/0.058) | F0–F2 | 0.0367 (0.0359/0.0376) |
| Gene Name | Comparison | Associated Function |
|---|---|---|
| wars1 | F0–F1 | growth/body shape/body size |
| tbx15 | F0–F1 | growth/body shape/body size |
| spry2 | F0–F1 | growth/body shape/body size |
| cast | F1–F2 | growth/body shape/body size |
| mef2b | F1–F2 | growth/body shape/body size |
| dock1 | F1–F2 | growth/body shape/body size |
| dock5 | F1–F2 | growth/body shape/body size |
| rnpc3 | F0–F2 | growth/body shape/body size |
| cntnap3 | F0–F1 | behaviour/nervous system |
| epha6 | F1–F2 | behaviour/nervous system |
| fmn1 | F1–F2 | behaviour/nervous system |
| dyrk1ab | F1–F2 | behaviour/nervous system |
| p2ry1 | F1–F2 | behaviour/nervous system |
| zmz1 | F1–F2 | behaviour/nervous system |
| mdga1 | F0–F2 | behaviour/nervous system |
| ephb2a | F1–F2 | immune response |
| ftr67 | F1–F2 | immune response |
| irf2bp1 | F1–F2 | immune response |
| arl16 | F1–F2 | immune response |
| lxn1 | F1–F2 | tumour suppression |
| emilin2 | F1–F2 | tumour suppression |
| Map2k4a | F1–F2, F0–F2 | tumour suppression |
| Map2k4b | F1–F2, F0–F2 | tumour suppression |
| ext1a | F0–F2 | tumour suppression |
| ext1b | F0–F2 | tumour suppression |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baesjou, J.-P.; Wellenreuther, M. Genomic Signatures of Domestication Selection in the Australasian Snapper (Chrysophrys auratus). Genes 2021, 12, 1737. https://doi.org/10.3390/genes12111737
Baesjou J-P, Wellenreuther M. Genomic Signatures of Domestication Selection in the Australasian Snapper (Chrysophrys auratus). Genes. 2021; 12(11):1737. https://doi.org/10.3390/genes12111737
Chicago/Turabian StyleBaesjou, Jean-Paul, and Maren Wellenreuther. 2021. "Genomic Signatures of Domestication Selection in the Australasian Snapper (Chrysophrys auratus)" Genes 12, no. 11: 1737. https://doi.org/10.3390/genes12111737