
Prenatal Diagnosis of Combined Maternal 4q Interstitial Deletion and Paternal 15q Microduplication

 , ,
, , 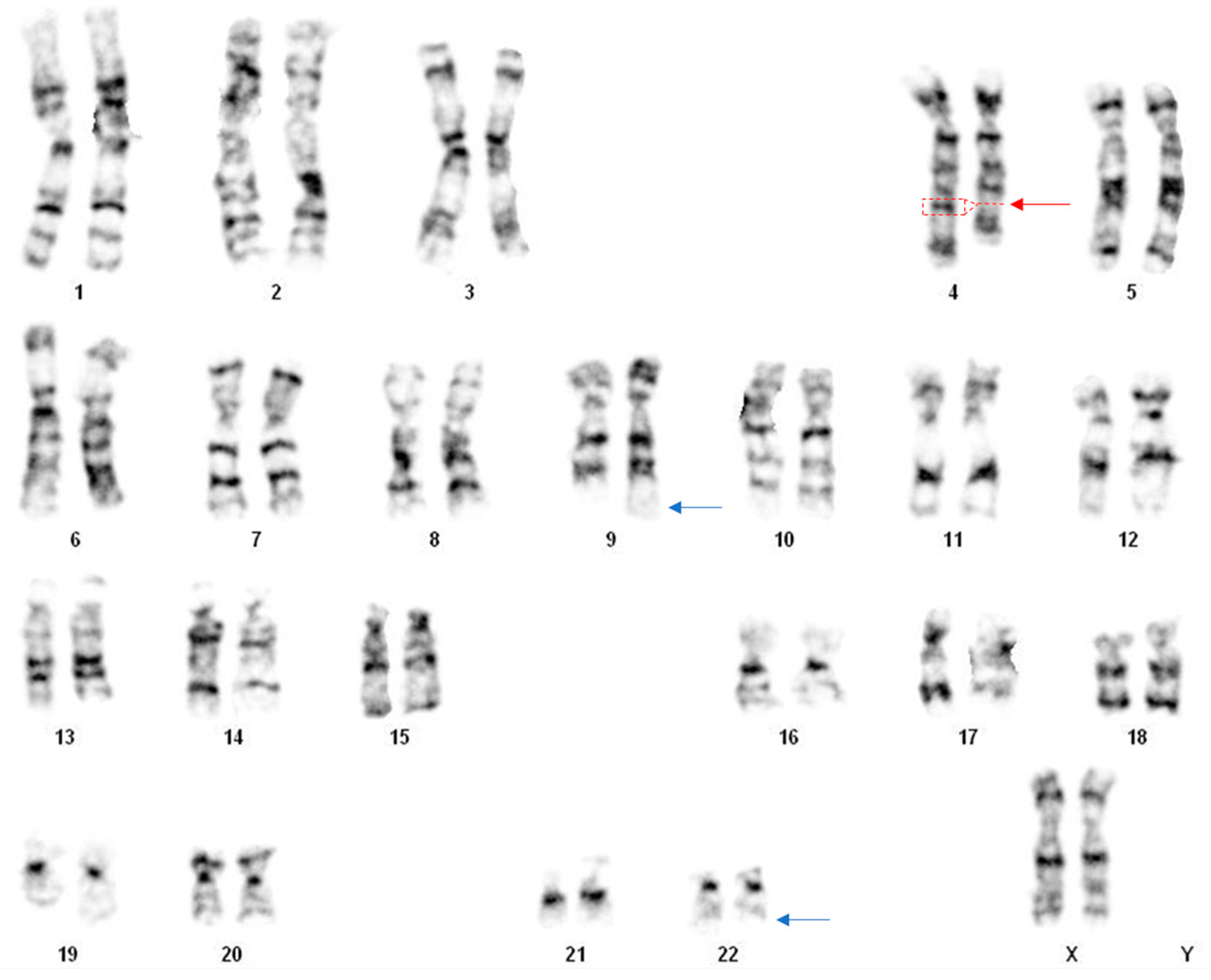{kind=link}
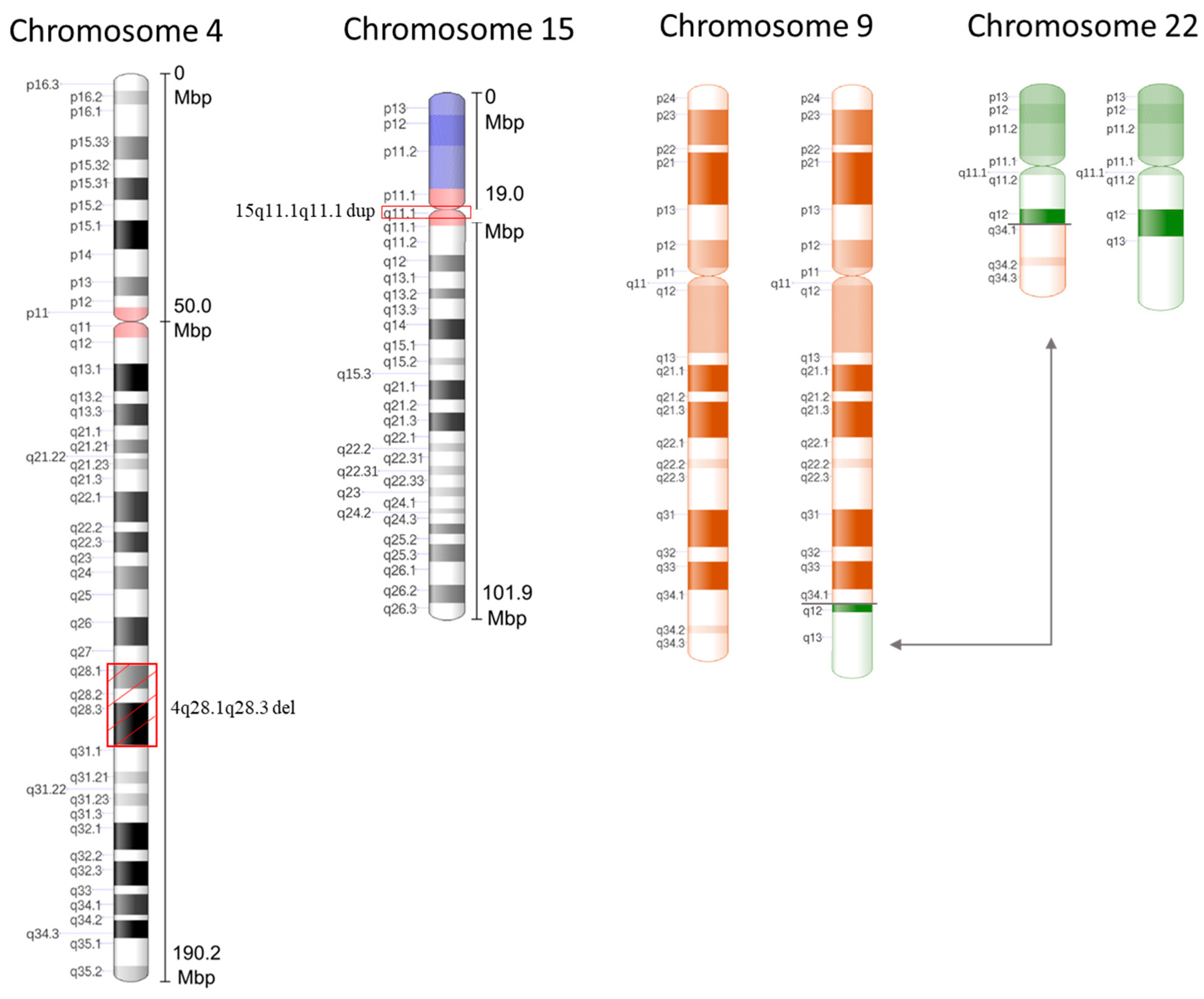{kind=link}
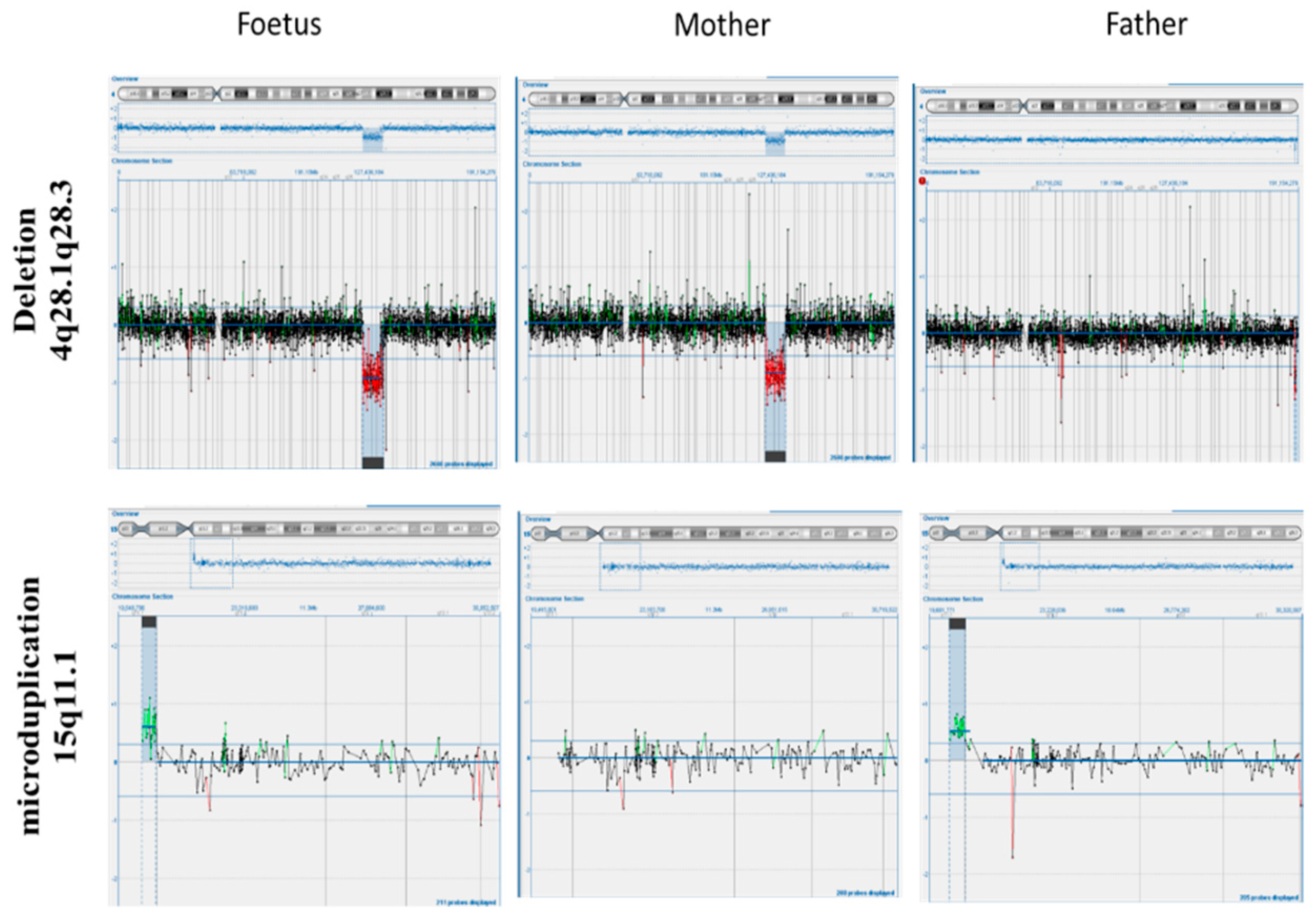{kind=link}
Abstract
:1. Introduction
2. Case Presentation
3. Materials and Methods
4. Discussion/Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Strehle, E.M.; Bantock, H.M. The Phenotype of Patients with 4q-Syndrome. Genet. Couns. 2003, 14, 195–205. [Google Scholar]
- Townes, P.L.; White, M.; Di Marzo, S. V 4q- Syndrome. Arch. Pediatrics Adolesc. Med. 1979, 133, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Dorner, S.; Stipoljev, F.; Skrlec, I.; Lauc, G.; Weise, A.; Mrasek, K.; Liehr, T.; Brecevic, L. Partial monosomy 4q and partial trisomy 13q: Phenotype and molecular mapping of the breakpoints. Chromosome Res. 2009, 17. [Google Scholar]
- Lin, A.E.; Garver, K.L.; Diggans, G.; Clemens, M.; Wenger, S.L.; Steele, M.W.; Jones, M.C.; Israel, J. Interstitial and Terminal Deletions of the Long Arm of Chromosome 4: Further Delineation of Phenotypes. Am. J. Med. Genet. 1988, 31, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Ismaila, S.; Helmy, N.A.; Mahmoud, W.M.; El-Ruby, M.O. Phenotypic Characterization of Rare Interstitial Deletion of Chromosome 4. J. Pediatric Genet. 2012, 1, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolakos, E.; Kefalas, K.; Vetro, A.; Oikonomidou, E.; Daskalakis, G.; Psara, N.; Siomou, E.; Papageorgiou, E.; Sevastopoulou, E.; Konstantinidou, A.; et al. Prenatal Diagnosis of Two de Novo 4q35-Qter Deletions Characterized by Array-CGH. Mol. Cytogenet. 2013, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Veyver, I.B.; Beaudet, A.L. Comparative Genomic Hybridization and Prenatal Diagnosis. Curr. Opin. Obstet. Gynecol. 2006, 18, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Black, J.; Kisiel, N.; Kulesz-Martin, M.F. SPAF, a New AAA-Protein Specific to Early Spermatogenesis and Malignant Conversion. Oncogene 2000, 19, 1579–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, A.J.; Cho, M.T.; Millan, F.; Juusola, J.; Retterer, K.; Joshi, C.; Niyazov, D.; Garnica, A.; Gratz, E.; Deardorff, M.; et al. Mutations in SPATA5 Are Associated with Microcephaly, Intellectual Disability, Seizures, and Hearing Loss. Am. J. Hum. Genet. 2015, 97, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Russell, Z.; Kontopoulos, E.V.; Quintero, R.A.; Debauche, D.M.; Ranells, J.D. Prenatal Diagnosis of a 4q33-4qter Deletion in a Fetus with Hydrops. Fetal Diagn. Ther. 2008, 24, 250–253. [Google Scholar] [CrossRef]
- Strehle, E.M.; Gruszfeld, D.; Schenk, D.; Mehta, S.G.; Simonic, I.; Huang, T. The Spectrum of 4q- Syndrome Illustrated by a Case Series. Gene 2012, 506, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Puusepp, S.; Kovacs-Nagy, R.; Alhaddad, B.; Braunisch, M.; Hoffmann, G.F.; Kotzaeridou, U.; Lichvarova, L.; Liiv, M.; Makowski, C.; Mandel, M.; et al. Compound Heterozygous SPATA5 Variants in Four Families and Functional Studies of SPATA5 Deficiency. Eur. J. Hum. Genet. 2018, 26, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Cappello, S.; Gray, M.J.; Badouel, C.; Lange, S.; Einsiedler, M.; Srour, M.; Chitayat, D.; Hamdan, F.F.; Jenkins, Z.A.; Morgan, T.; et al. Mutations in Genes Encoding the Cadherin Receptor-Ligand Pair DCHS1 and FAT4 Disrupt Cerebral Cortical Development. Nat. Genet. 2013, 45, 1300–1308. [Google Scholar] [CrossRef]
- Wang, S.; Liu, A.; Wu, G.; Ding, H.F.; Huang, S.; Nahman, S.; Dong, Z. The CPLANE Protein Intu Protects Kidneys from Ischemia-Reperfusion Injury by Targeting STAT1 for Degradation. Nat. Commun. 2018, 9, 1234. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.J. Molecular Networking in the Neuronal Ceroid Lipofuscinoses: Insights from Mammalian Models and the Social Amoeba Dictyostelium Discoideum. J. Biomed. Sci. 2020, 27, 1–16. [Google Scholar]
- Yamamoto, S.; Kitagawa, D. Self-Organization of Plk4 Regulates Symmetry Breaking in Centriole Duplication. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Wang, Y.; Wen, S.W.; Zhao, X.; Hu, W.; Liu, C.; Gao, L.; Zhang, Y.; Wang, S.; Yang, X.; et al. Recombinant chromosome 4 in two fetuses-case report and literature review. Mol. Cytogenet. 2018, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Tidrenczel, Z.; Tardy, E.P.; Pikó, H.; et al. Prenatal diagnosis of 4q terminal deletion and review of the literature. Cytogenet. Genome Res. 2019, 158, 63–73. [Google Scholar] [CrossRef]
- Vlaikou, A.M.; Manolakos, E.; Noutsopoulos, D.; Sarkadi, E.; Böjtös, I.; Demeter, J.; Simon, J.; Kósa, J.P.; Beke, A. An interstitial 4q31. 21q31. 22 microdeletion associated with developmental delay: Case report and literature review. Cytogenet. Genome Res. 2014, 142, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Strehle, E.M.; Yu, L.; Rosenfeld, J.A.; et al. Genotype–phenotype analysis of 4q deletion syndrome: Proposal of a critical region. Am. J. Med. Genet. A 2012, 158, 2139–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Sun, Y.; Huo, P.; Tian, H.; Gao, J.; Li, Y. Prenatal diagnosis of a maternal 7.22-Mb deletion at chromosome 4q32.2q32.3 by SNP array. Mol. Cytogenet. 2020, 13, 12. [Google Scholar] [CrossRef]
- Aladhami, S.M.; Gould, C.P.; Muhammad, F.A. A new inherited interstitial deletion of the distal long arm of chromosome 4. Hum. Hered. 2000, 50, 146–150. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Libotte, F.; Fabiani, M.; Margiotti, K.; Viola, A.; Mesoraca, A.; Giorlandino, C. Prenatal Diagnosis of Combined Maternal 4q Interstitial Deletion and Paternal 15q Microduplication. Genes 2021, 12, 1626. https://doi.org/10.3390/genes12101626
Libotte F, Fabiani M, Margiotti K, Viola A, Mesoraca A, Giorlandino C. Prenatal Diagnosis of Combined Maternal 4q Interstitial Deletion and Paternal 15q Microduplication. Genes. 2021; 12(10):1626. https://doi.org/10.3390/genes12101626
Chicago/Turabian StyleLibotte, Francesco, Marco Fabiani, Katia Margiotti, Antonella Viola, Alvaro Mesoraca, and Claudio Giorlandino. 2021. "Prenatal Diagnosis of Combined Maternal 4q Interstitial Deletion and Paternal 15q Microduplication" Genes 12, no. 10: 1626. https://doi.org/10.3390/genes12101626