
WDR36-Associated Neurodegeneration: A Case Report Highlights Possible Mechanisms of Normal Tension Glaucoma

Abstract
:1. Introduction
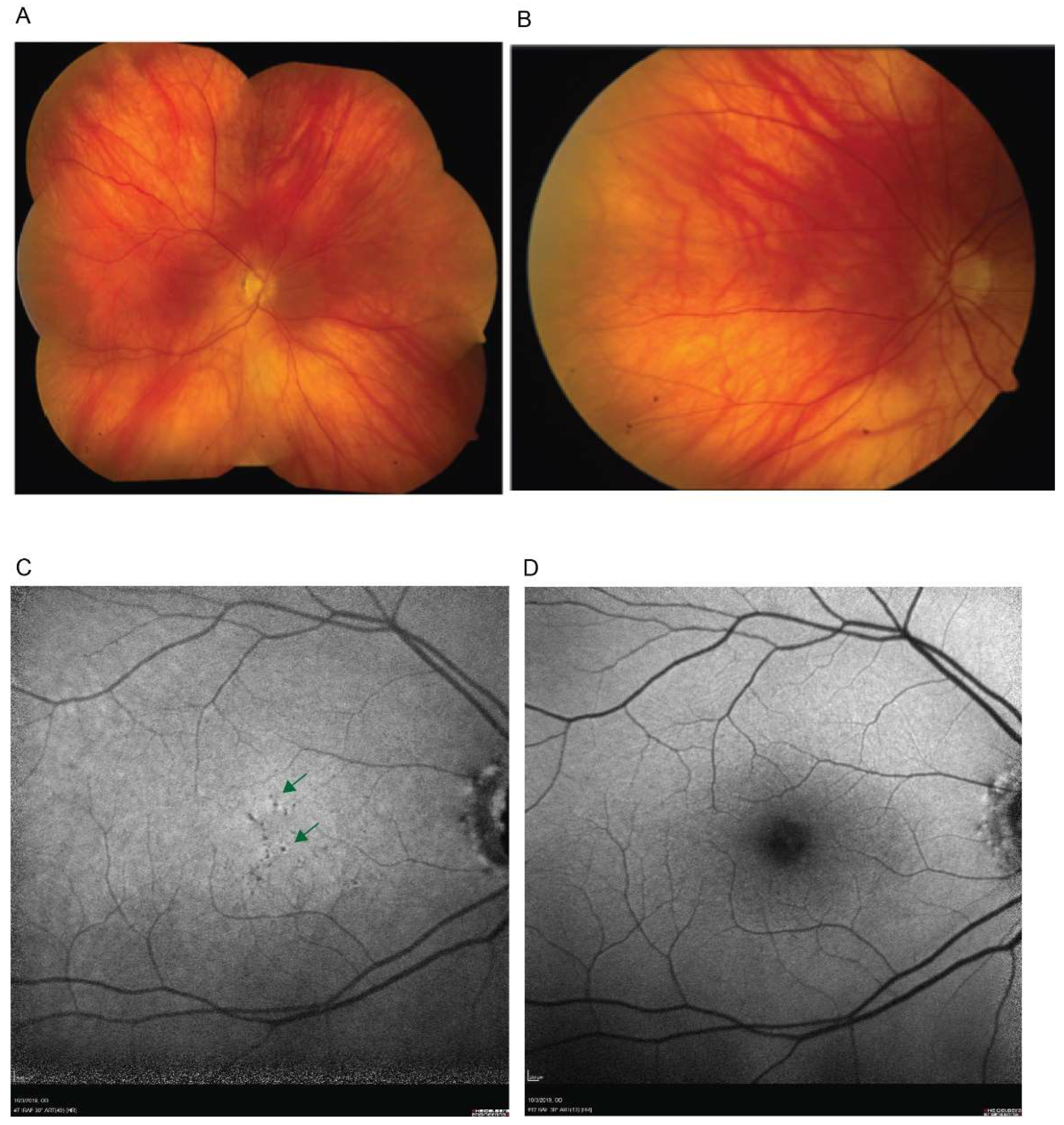
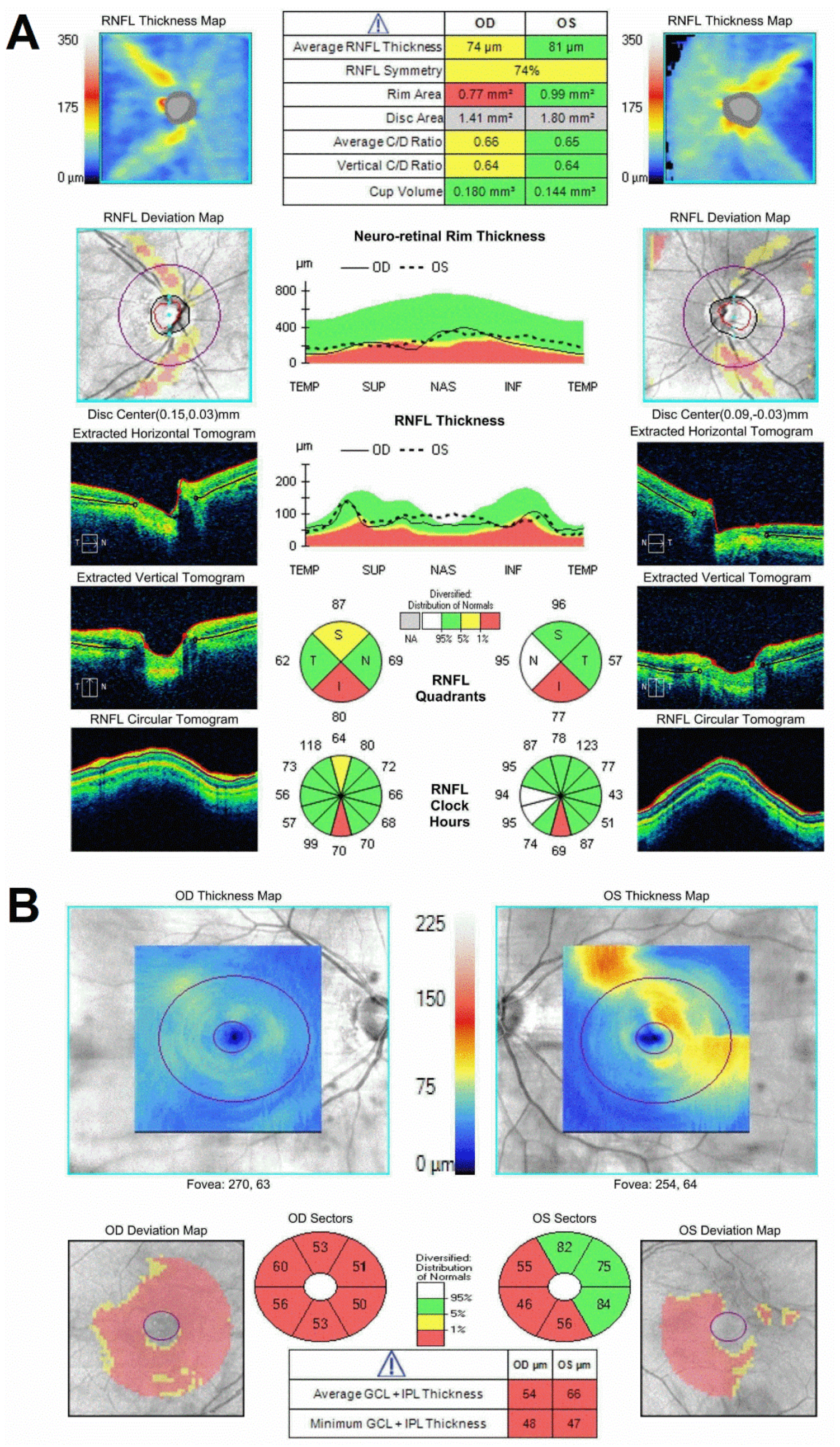
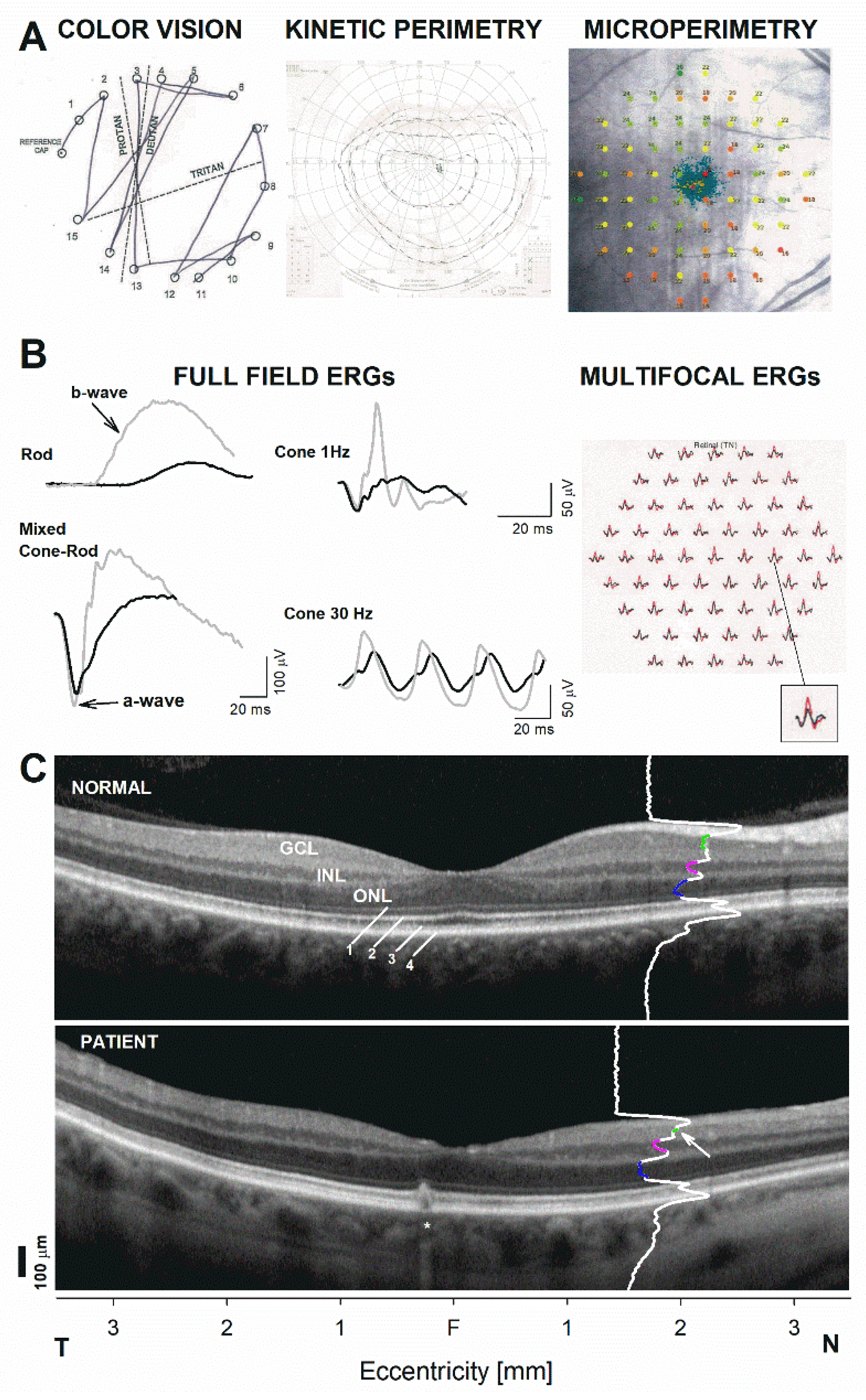
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R. Glaucoma in developing countries. Indian J. Ophthalmol. 2012, 60, 446–450. [Google Scholar] [CrossRef]
- Jonas, J.B.; Aung, T.; Bourne, R.R.; Bron, A.M.; Ritch, R.; Panda-Jonas, S. Glaucoma. Lancet 2017, 390, 2183–2193. [Google Scholar] [CrossRef]
- Bertaud, S.; Aragno, V.; Baudouin, C.; Labbe, A. Primary open-angle glaucoma. Rev. Med. Interne 2019, 40, 445–452. [Google Scholar] [CrossRef]
- Mansouri, K.; Medeiros, F.A.; Weinreb, R.N. Global rates of glaucoma surgery. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 2609–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallick, J.; Devi, L.; Malik, P.K.; Mallick, J. Update on Normal Tension Glaucoma. J. Ophthalmic Vis. Res. 2016, 11, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Hauser Michael, A.; Sena, D.F.; Flor, J.; Walter, J.; Auguste, J.; LaRocque-Abramson, K.; Graham, F.; DelBono, E.; Haines, J.L.; Pericak-Vance, M.A.; et al. Distribution of optineurin sequence variations in an ethnically diverse population of low-tension glaucoma patients from the United States. J. Glaucoma 2006, 15, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Wiggs, J.L.; Pasquale, L.R. Genetics of glaucoma. Hum. Mol. Genet. 2017, 26, R21–R27. [Google Scholar] [CrossRef] [PubMed]
- Gharahkhani, P.; Burdon, K.P.; Fogarty, R.; Sharma, S.; Hewitt, A.W.; Martin, S.; Law, M.H.; Cremin, K.; Cooke Bailey, J.N.; Loomis, S.J.; et al. Common variants near ABCA1, AFAP1 and GMDS confer risk of primary open-angle glaucoma. Nat. Genet. 2014, 46, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.W.; Gudiseva, H.V.; Chavali, V.R.M.; Trachtman, B.; Ramakrishnan, M.; Merritt, W.T., III; Pistilli, M.; Rossi, R.A.; Blachon, S.; Sankar, P.S.; et al. The MT-CO1 V83I Polymorphism is a Risk Factor for Primary Open-Angle Glaucoma in African American Men. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1751–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudiseva, H.V.; Pistilli, M.; Salowe, R.; Singh, L.N.; Collins, D.W.; Cole, B.; He, J.; Merriam, S.; Khachataryan, N.; Henderer, J.; et al. The association of mitochondrial DNA haplogroups with POAG in African Americans. Exp. Eye Res. 2019, 181, 85–89. [Google Scholar] [CrossRef]
- Chi, Z.-L.; Yasumoto, F.; Sergeev, Y.; Minami, M.; Obazawa, M.; Kimura, I.; Takada, Y.; Iwata, T. Mutant WDR36 directly affects axon growth of retinal ganglion cells leading to progressive retinal degeneration in mice. Hum. Mol. Genet. 2010, 19, 3806–3815. [Google Scholar] [CrossRef] [Green Version]
- Sihota, R.; Angmo, D.; Ramaswamy, D.; Dada, T. Simplifying “target” intraocular pressure for different stages of primary open-angle glaucoma and primary angle-closure glaucoma. Indian J. Ophthalmol. 2018, 66, 495–505. [Google Scholar] [CrossRef]
- Skarie, J.M.; Link, B.A. The primary open-angle glaucoma gene WDR36 functions in ribosomal RNA processing and interacts with the p53 stress-response pathway. Hum. Mol. Genet. 2008, 17, 2474–2485. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.J.; Wang, D.Y.; Cheng, C.Y.; Ko, W.C.; Lam, S.C.; Pang, C.P. Different WDR36 mutation pattern in Chinese patients with primary open-angle glaucoma. Mol. Vis. 2009, 15, 646–653. [Google Scholar]
- Dores, G.M.; Curtis, R.E.; Toro, J.R.; Devesa, S.S.; Fraumeni, J.F., Jr. Incidence of cutaneous sebaceous carcinoma and risk of associated neoplasms: Insight into Muir-Torre syndrome. Cancer 2008, 113, 3372–3381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuse, N. Genetic bases for glaucoma. Tohoku J. Exp. Med. 2010, 221, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, M.A.; Allingham, R.R.; Linkroum, K.; Wang, J.; RaRocque-Abramson, K.; Figueiredo, D.; Santiago-Turla, C.; del Bono, E.A.; Haines, J.L.; Pericak-Vance, M.A.; et al. Distribution of WDR36 DNA sequence variants in patients with primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2542–2546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, K.A.; Gallagher, J.E.; Mitchell, B.M.; Granneman, S.; Baserga, S.J. The small-subunit processome is a ribosome assembly intermediate. Eukaryot Cell 2004, 3, 1619–1626. [Google Scholar] [CrossRef] [Green Version]
- Gallenberger, M.; Meinel, D.M.; Kroeber, M.; Wegner, M.; Milkereit, P.; Bösl, M.R.; Tamm, E.R. Lack of WDR36 leads to preimplantation embryonic lethality in mice and delays the formation of small subunit ribosomal RNA in human cells in vitro. Hum. Mol. Genet. 2011, 20, 422–435. [Google Scholar] [CrossRef] [Green Version]
- Monemi, S.; Spaeth, G.; DaSilva, A.; Popinchalk, S.; Ilitchev, E.; Liebmann, J.; Ritch, R.; Héon, E.; Crick, R.P.; Child, A.; et al. Identification of a novel adult-onset primary open-angle glaucoma (POAG) gene on 5q22.1. Hum. Mol. Genet. 2005, 14, 725–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, M.; Bierry, M.C.; Kobayashi, S.V.; Ward, T.; Schimmack, G.; Burchard, J.; Schelter, J.M.; Dai, H.; He, Y.D.; Linsley, P.S. T lymphocyte activation gene identification by coregulated expression on DNA microarrays. Genomics 2004, 83, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Motushchuk, A.E.; Komarova, T.Y.; Grudinina, N.A.; Rakhmanov, V.V.; Mandelshtam, M.Y.; Astakhov, Y.S.; Vasilyev, V.B. Genetic variants of CYP1B1 and WDR36 in the patients with primary congenital glaucoma and primary open angle glaucoma from Saint-Petersburg. Genetika 2009, 45, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.-Y.; Tam, P.O.-P.; Chiang, S.W.-Y.; Ding, N.; Cheon, L.J.; Yam, G.H.-F.; Pang, C.-P.; Wang, N.-L. Multiple gene polymorphisms analysis revealed a different profile of genetic polymorphisms of primary open-angle glaucoma in northern Chinese. Mol. Vis. 2009, 15, 89–98. [Google Scholar] [PubMed]
- Pasutto, F.; Mardin, C.Y.; Michels-Rautenstrauss, K.; Weber, B.H.F.; Sticht, H.; Chavarria-Soley, G.; Rautenstrauss, B.; Kruse, F.; Reis, A. Profiling of WDR36 missense variants in German patients with glaucoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 270–274. [Google Scholar] [CrossRef] [Green Version]
- Fingert, J.H.; Alward, W.L.M.; Kwon, Y.H.; Shanka, S.P.; Andorf, J.L.; Mackey, D.A.; Scheffield, V.C.; Stone, E.M. No association between variations in the WDR36 gene and primary open-angle glaucoma. Arch. Ophthalmol. 2007, 125, 434–436. [Google Scholar] [CrossRef]
- Footz, T.K.; Johnson, J.L.; Dubois, S.; Boivin, N.; Raymond, V.; Walter, M.A. Glaucoma-associated WDR36 variants encode functional defects in a yeast model system. Hum. Mol. Genet. 2009, 18, 1276–1287. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, A.W.; Dimasi, D.P.; Mackey, D.A.; Craig, J.E. A Glaucoma Case-control Study of the WDR36 Gene D658G sequence variant. Am. J. Ophthalmol. 2006, 142, 324–325. [Google Scholar] [CrossRef]
- Weisschuh, N.; Wolf, C.; Wissinger, B.; Gramer, E. Variations in the WDR36 gene in German patients with normal tension glaucoma. Mol. Vis. 2007, 13, 724–729. [Google Scholar]
- Kramer, P.L.; Samples, J.R.; Monemi, S.; Sykes, R.; Sarfarazi, M.; Wirtz, M.K. The role of the WDR36 gene on chromosome 5q22.1 in a large family with primary open-angle glaucoma mapped to this region. Arch. Ophthalmol. 2006, 124, 1328–1331. [Google Scholar] [CrossRef] [Green Version]
- Frezzotti, P.; Pescucci, C.; Papa, F.T.; Iester, M.; Mittica, V.; Motolese, I.; Peruzzi, S.; Artuso, R.; Longo, I.; Mencarelli, M.A.; et al. Association between primary open-angle glaucoma (POAG) and WDR36 sequence variance in Italian families affected by POAG. Br. J. Ophthalmol. 2011, 95, 624–626. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; He, W.; Zhao, J.; Zeng, Y.; Cheng, H. Association of WDR36 polymorphisms with primary open angle glaucoma: A systematic review and meta-analysis. Medicine 2017, 96, e7291. [Google Scholar] [CrossRef]
- Blanco-Marchite, C.; Sánchez-Sánchez, F.; López-Garrido, M.-P.; Iñigez-de-Onzoño, M.; López-Martínez, F.; López-Sánchez, E.; Alvarez, L.; Rodríguez-Calvo, P.-P.; Méndez-Hernández, C.; Fernández-Vega, L.; et al. WDR36 and P53 gene variants and susceptibility to primary open-angle glaucoma: Analysis of gene-gene interactions. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8467–8478. [Google Scholar] [CrossRef]
- Miyazawa, A.; Fuse, N.; Mengkegale, M.; Ryu, M.; Seimiya, M.; Wada, Y.; Nishida, K. Association between primary open-angle glaucoma and WDR36 DNA sequence variants in Japanese. Mol. Vis. 2007, 13, 1912–1919. [Google Scholar] [PubMed]
- Huang, X.; Li, M.; Guo, X.; Li, S.; Xiao, X.; Jia, X.; Liu, X.; Zhang, Q. Mutation analysis of seven known glaucoma-associated genes in Chinese patients with glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3594–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, C.P.; Fan, B.J.; Canlas, O.; Wang, D.Y.; Dubois, S.; Tam, P.O.S.; Lam, D.S.C.; Raymond, V.; Ritch, R. A genome-wide scan maps a novel juvenile-onset primary open angle glaucoma locus to chromosome 5q. Mol. Vis. 2006, 12, 85–92. [Google Scholar] [PubMed]
- Mookherjee, S.; Chakraborty, S.; Vishal, M.; Banerjee, D.; Sen, A.; Ray, K. WDR36 variants in East Indian primary open-angle glaucoma patients. Mol. Vis. 2011, 17, 2618–2627. [Google Scholar] [PubMed]
- Williams, S.E.; Carmichael, T.R.; Allingham, R.R.; Hauser, M.; Ramsay, M. The genetics of POAG in black South Africans: A candidate gene association study. Sci Rep. 2015, 5, 8378. [Google Scholar] [CrossRef] [PubMed]
- Blankenbach, K.V.; Bruno, G.; Wondra, E.; Spohner, A.K.; Aster, N.J.; Vienken, H.; Trautmann, S.; Ferreirós, N.; Wieland, T.; Bruni, P.; et al. The WD40 repeat protein, WDR36, orchestrates sphingosine kinase-1 recruitment and phospholipase C-β activation by Gq-coupled receptors. Biochim. Biophys Acta Mol. Cell Biol. Lipids 2020, 1865, 158704. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Lab Test Name | Normal Values | Patient Values |
|---|---|---|
| C Reactive Protein (CRP) | <8.0 mg/L | 6.2 mg/L |
| B12 | 160–950 pg/mL | 812 pg/mL |
| Folate | 2.7–17.0 ng/mL | 16.8 ng/mL |
| Methylmalonic acid | 87–318 nmol/L | 251 nmol/L |
| Homocysteine | <11.4 μmol/L | 11.4 μmol/L |
| OPA1 | Associated mutation 2826delT | No mutations |
| LHON | Associated mutations (mt.3460G > A, mt.11778G > A, and mt.14484T > C) | No mutations |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meer, E.; Aleman, T.S.; Ross, A.G. WDR36-Associated Neurodegeneration: A Case Report Highlights Possible Mechanisms of Normal Tension Glaucoma. Genes 2021, 12, 1624. https://doi.org/10.3390/genes12101624
Meer E, Aleman TS, Ross AG. WDR36-Associated Neurodegeneration: A Case Report Highlights Possible Mechanisms of Normal Tension Glaucoma. Genes. 2021; 12(10):1624. https://doi.org/10.3390/genes12101624
Chicago/Turabian StyleMeer, Elana, Tomas S. Aleman, and Ahmara G. Ross. 2021. "WDR36-Associated Neurodegeneration: A Case Report Highlights Possible Mechanisms of Normal Tension Glaucoma" Genes 12, no. 10: 1624. https://doi.org/10.3390/genes12101624