A Transcriptome Analysis Identifies Biological Pathways and Candidate Genes for Feed Efficiency in DLY Pigs

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Tissues
2.3. RNA Preparation
2.4. RNA Sequencing
2.5. Quality Control, Mapping, and Quantification
2.6. Identification of DEGs
2.7. GO and KEGG Pathway Enrichment Analysis
2.8. Reactome Pathway Enrichment Analysis
2.9. PPI Network Construction
2.10. Validation of Differentially-Expressed Genes by RT-qPCR
2.11. Data Availability
3. Results
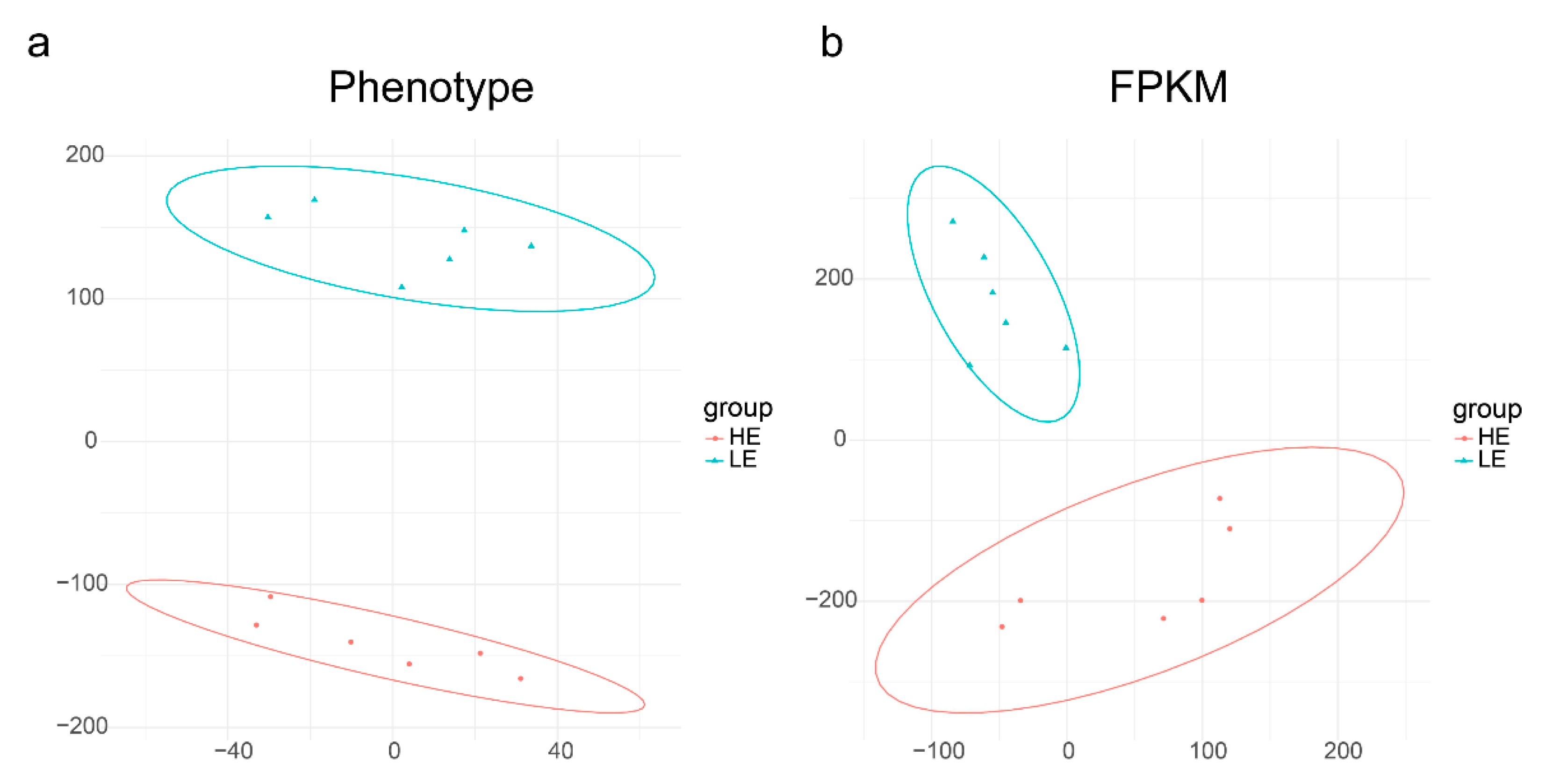
3.1. The Phenotype of the High and Low FE Groups Showed a Dramatic Difference
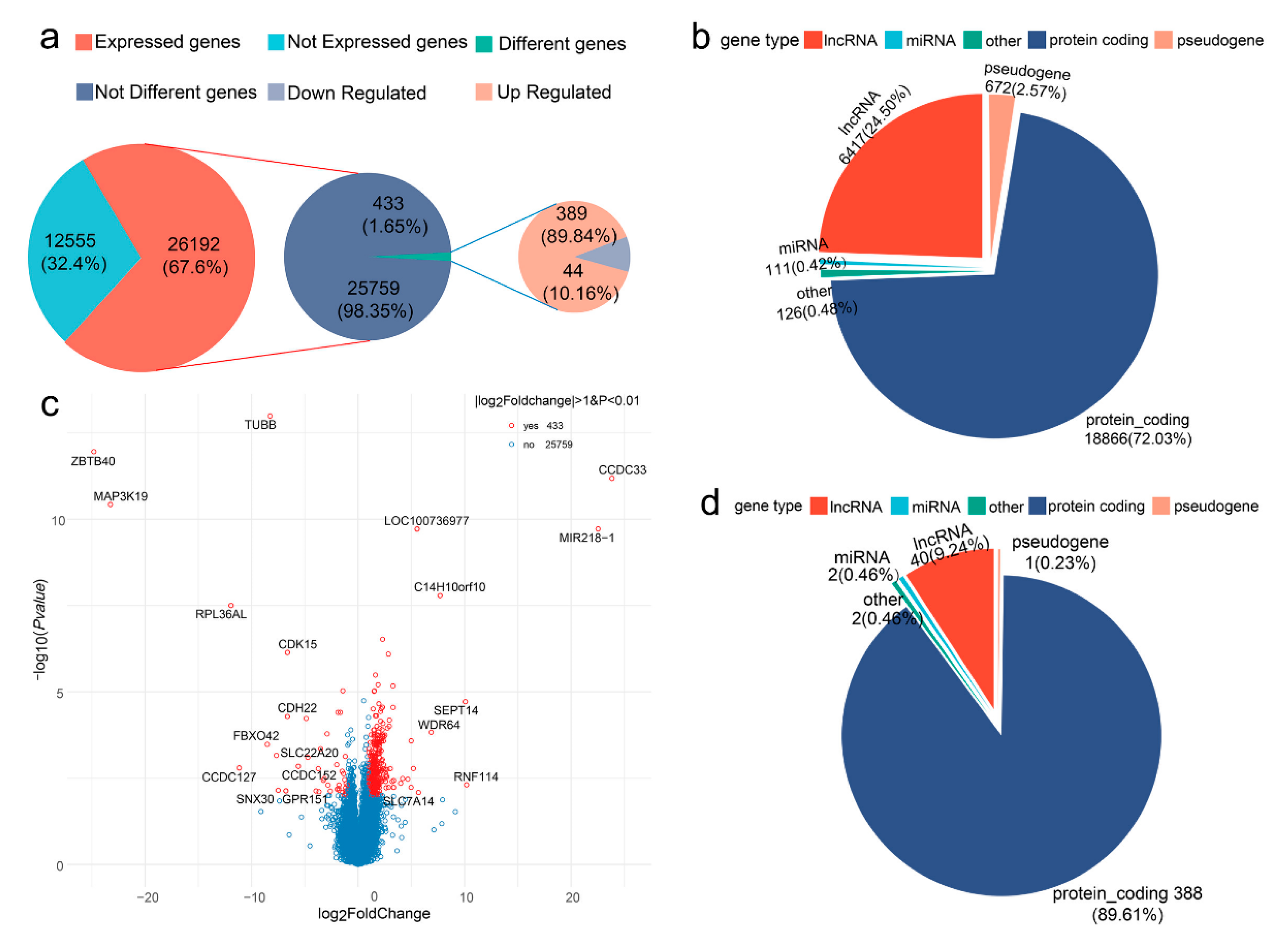
3.2. Summary of RNA-seq Data
3.3. Differentially Expressed Genes between Low and High FE Pigs
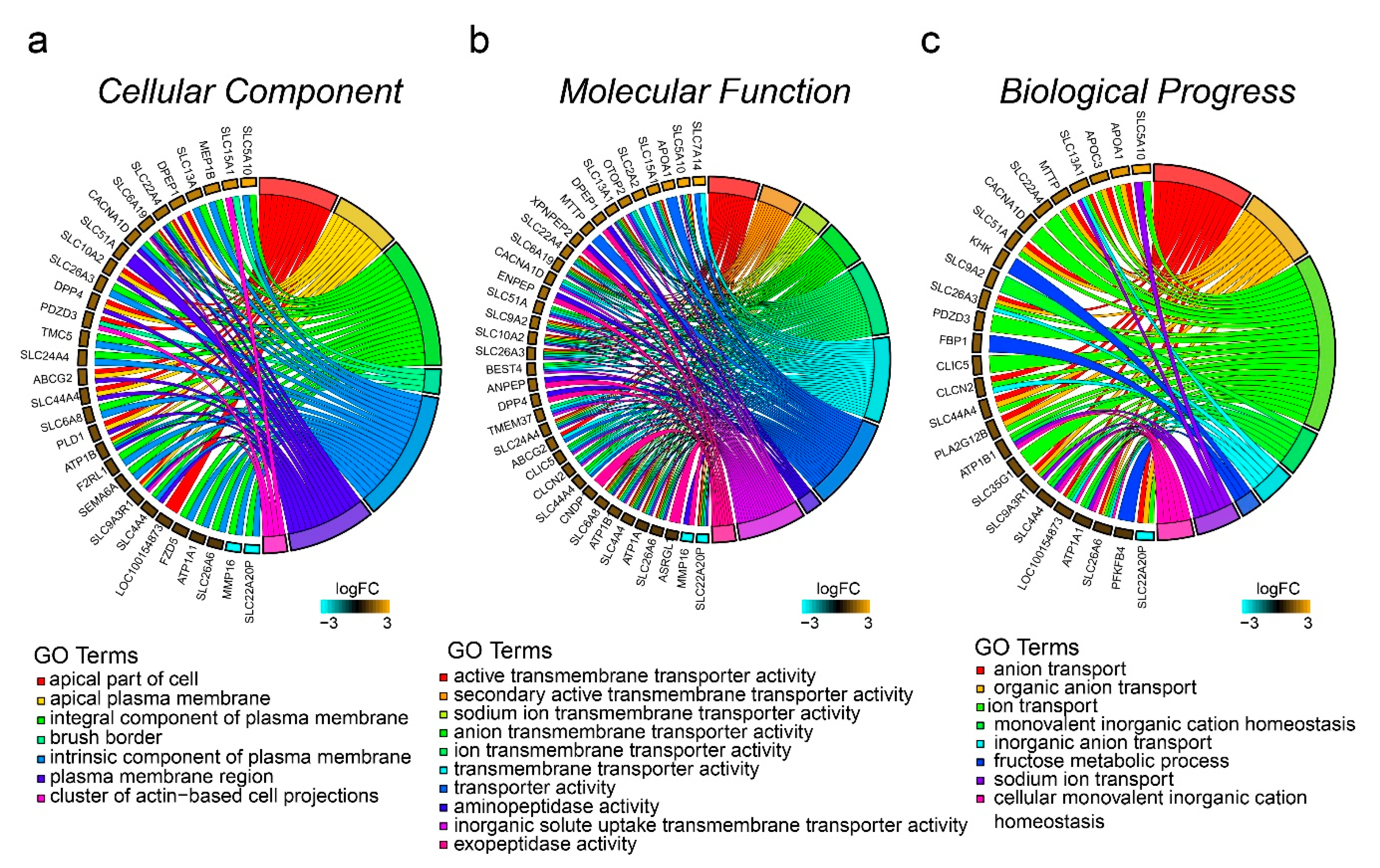
3.4. Functional Annotation Based on GO
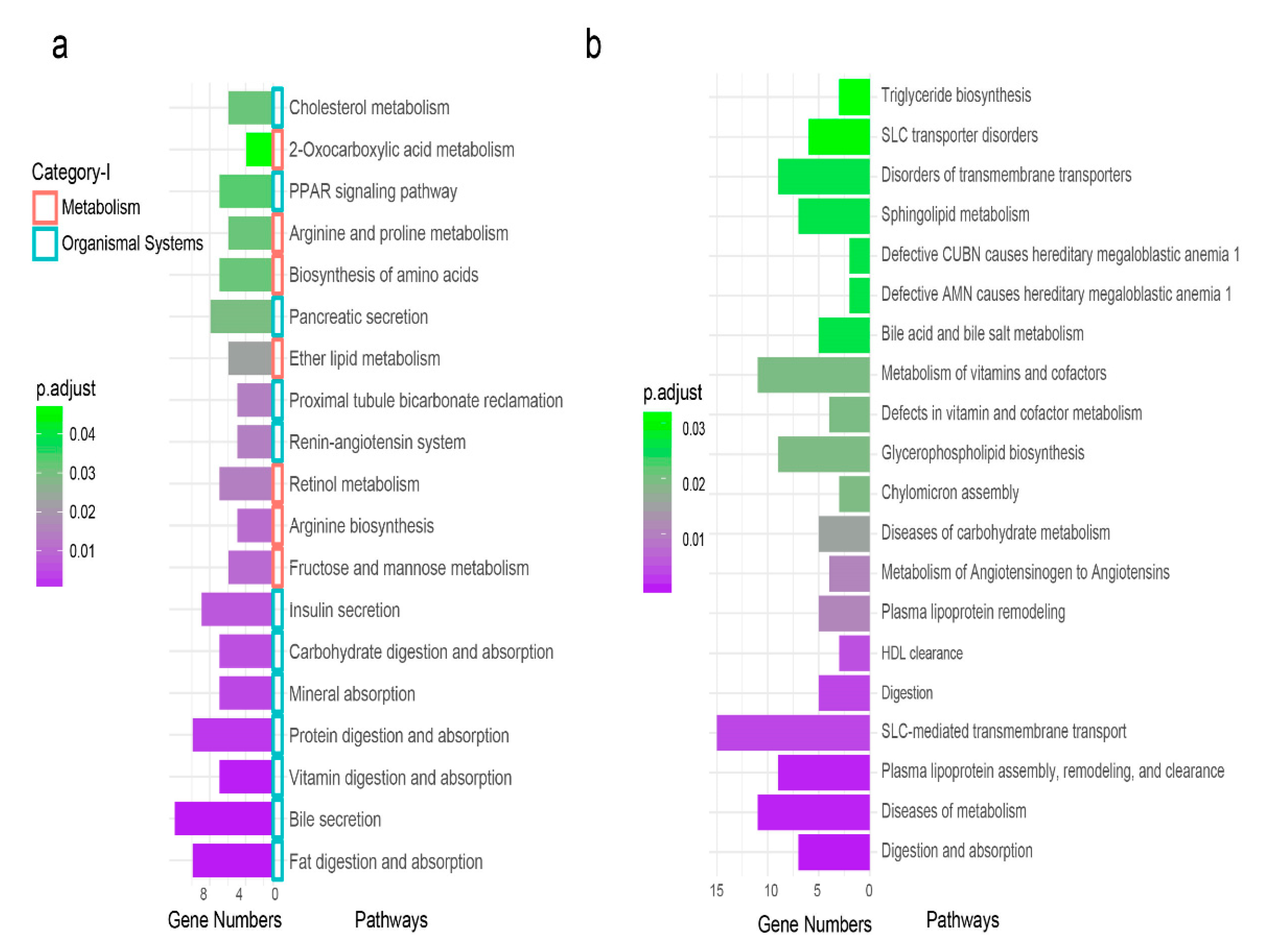
3.5. KEGG Functional Enrichment Pathways
3.6. Reactome Functional Enrichment Pathways
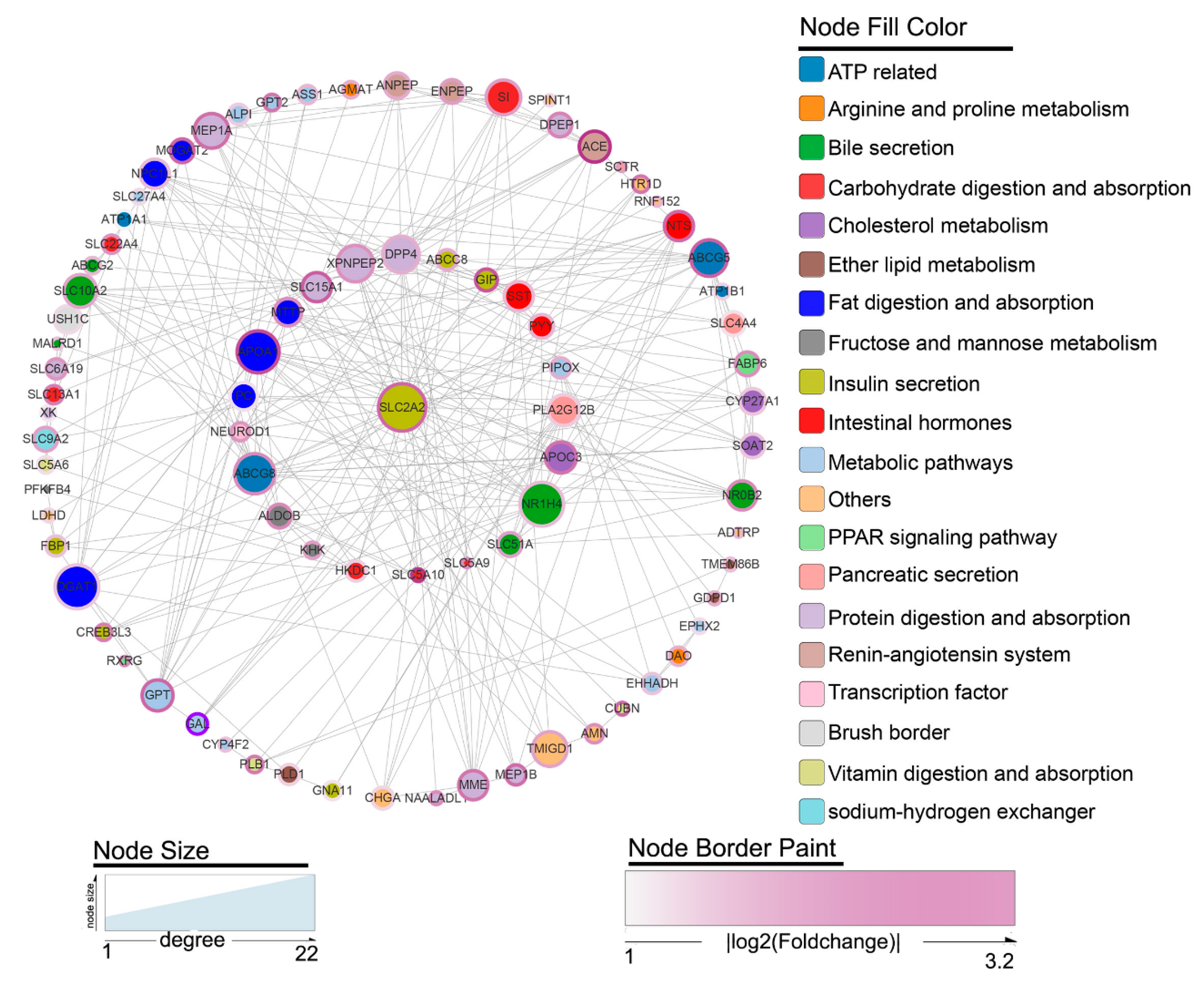
3.7. Protein-Protein Interaction (PPI) Network
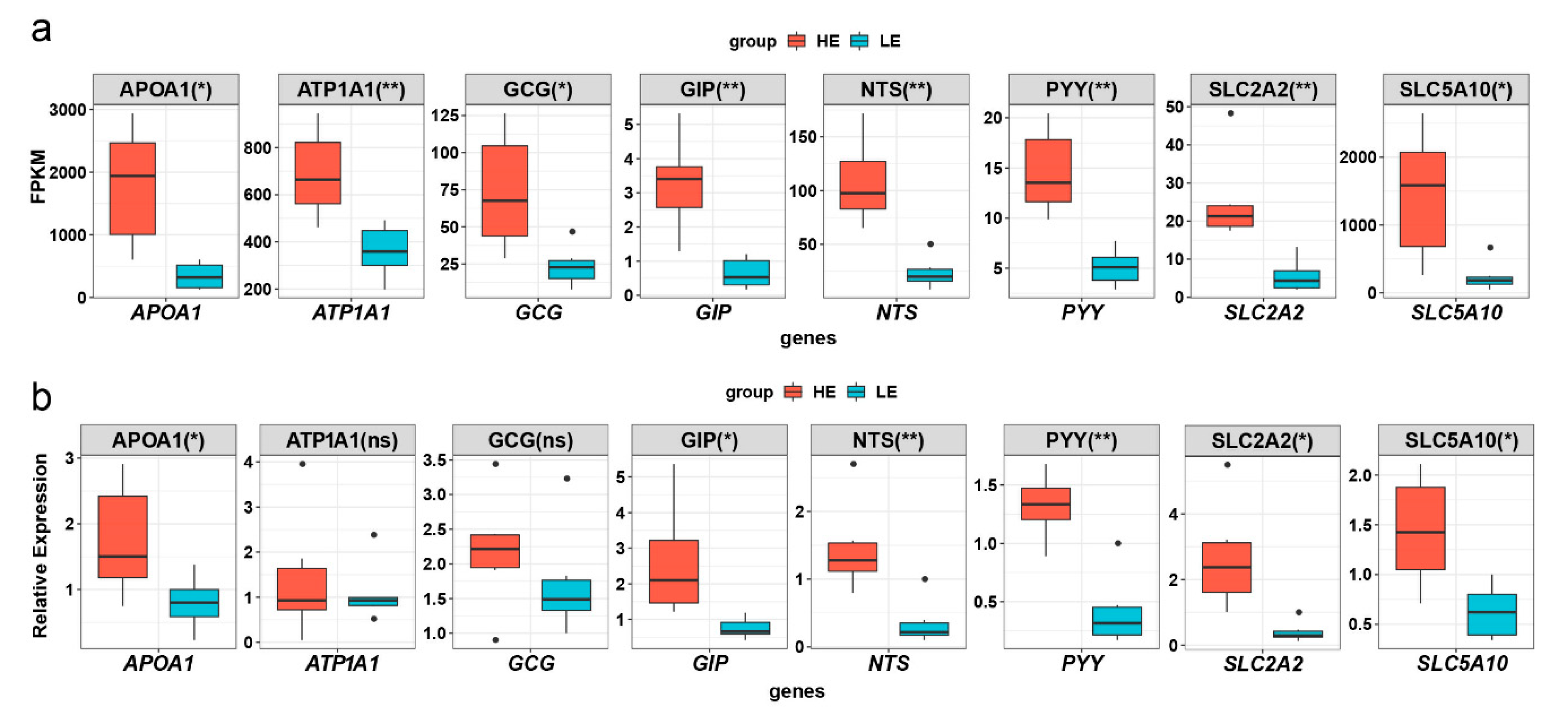
3.8. Quantitative Real Time PCR Validation of Eight DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hoque, M.A.; Kadowaki, H.; Shibata, T.; Oikawa, T.; Suzuki, K. Genetic parameters for measures of residual feed intake and growth traits in seven generations of Duroc pigs. Livest. Sci. 2009, 121, 45–49. [Google Scholar] [CrossRef]
- Van Heugten, E. Growing-Finishing Swine Nutrient Recommendations and Feeding Management; National Swine Nutrition Guide: Manhattan, KS, USA, 2010; pp. 80–95. [Google Scholar]
- Kanis, E.; De Greef, K.; Hiemstra, A.; Van Arendonk, J. Breeding for societally important traits in pigs. J. Anim. Sci. 2005, 83, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiong, X.; Yang, J.; Zhou, L.; Yang, B.; Ai, H.; Ma, H.; Xie, X.; Huang, Y.; Fang, S. Genome-wide association analyses for meat quality traits in Chinese Erhualian pigs and a Western Duroc×(Landrace× Yorkshire) commercial population. Genet. Sel. Evol. 2015, 47, 44. [Google Scholar] [CrossRef] [PubMed]
- World Food and Agriculture. FAO Statistical Yearbook; FAO: Rome, Italy, 2013. [Google Scholar]
- Koch, R.M.; Swiger, L.A.; Chambers, D.; Gregory, K.E. Efficiency of feed use in beef cattle. J. Anim. Sci. 1963, 22, 486–494. [Google Scholar] [CrossRef]
- Aggrey, S.E.; Karnuah, A.B.; Sebastian, B.; Anthony, N.B. Genetic properties of feed efficiency parameters in meat-type chickens. Genet. Sel. Evol. 2010, 42, 25. [Google Scholar] [CrossRef]
- Fix, J.; Cassady, J.; Van Heugten, E.; Hanson, D.; See, M. Differences in lean growth performance of pigs sampled from 1980 and 2005 commercial swine fed 1980 and 2005 representative feeding programs. Livest. Sci. 2010, 128, 108–114. [Google Scholar] [CrossRef]
- Cai, W.; Casey, D.S.; Dekkers, J.C.M. Selection response and genetic parameters for residual feed intake in Yorkshire swine. J. Anim. Sci. 2008, 86, 287–298. [Google Scholar] [CrossRef]
- Hoque, M.A.; Kadowaki, H.; Shibata, T.; Suzuki, K. Maternal and direct genetic parameters for production traits and maternal correlations among production and feed efficiency traits in Duroc pigs. J. Anim. Sci. 2008, 21, 961–966. [Google Scholar] [CrossRef]
- Do, D.N.; Strathe, A.B.; Jensen, J.; Mark, T.; Kadarmideen, H.N. Genetic parameters for different measures of feed efficiency and related traits in boars of three pig breeds. J. Anim. Sci. 2013, 91, 4069–4079. [Google Scholar] [CrossRef]
- Gilbert, H.; Bidanel, J.P.; Billon, Y.; Lagant, H.; Guillouet, P.; Sellier, P.; Noblet, J.; Hermesch, S. Correlated responses in sow appetite, residual feed intake, body composition, and reproduction after divergent selection for residual feed intake in the growing pig. J. Anim. Sci. 2012, 90, 1097–1108. [Google Scholar] [CrossRef]
- Nasiri Foomani, N.; Zerehdaran, S.; Ahani Azari, M.; Lotfi, E. Genetic parameters for feed efficiency and body weight traits in Japanese quail. Br. Poult. Sci. 2014, 55, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Do, D.N.; Ostersen, T.; Strathe, A.B.; Mark, T.; Jensen, J.; Kadarmideen, H.N. Genome-wide association and systems genetic analyses of residual feed intake, daily feed consumption, backfat and weight gain in pigs. BMC Genet. 2014, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Ren, J.; Ren, D.R.; Ma, J.W.; Guo, Y.M.; Huang, L.S. Mapping quantitative trait loci for feed consumption and feeding behaviors in a White Duroc x Chinese Erhualian resource population. J. Anim. Sci. 2009, 87, 3458–3463. [Google Scholar] [CrossRef] [PubMed]
- Sahana, G.; Kadlecova, V.; Hornshoj, H.; Nielsen, B.; Christensen, O.F. A genome-wide association scan in pig identifies novel regions associated with feed efficiency trait. J. Anim. Sci. 2013, 91, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Onteru, S.K.; Gorbach, D.M.; Young, J.M.; Garrick, D.J.; Dekkers, J.C.; Rothschild, M.F. Whole Genome Association Studies of Residual Feed Intake and Related Traits in the Pig. PLoS ONE 2013, 8, e61756. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Quan, J.; Yang, M.; Wang, X.; Zheng, E.; Yang, H.; Fu, D.; Yang, Y.; Yang, L.; Li, Z. Genome-wide association analysis reveals genetic loci and candidate genes for feeding behavior and eating efficiency in Duroc boars. PLoS ONE 2017, 12, e0183244. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Yang, M.; Wang, X.; Quan, J.; Zhuang, Z.; Zhou, S.; Li, S.; Xu, Z.; Zheng, E.; Cai, G. Genetic architecture of feeding behavior and feed efficiency in a Duroc pig population. Front. Genet. 2018, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- Do, D.N.; Strathe, A.B.; Ostersen, T.; Pant, S.D.; Kadarmideen, H.N. Genome-wide association and pathway analysis of feed efficiency in pigs reveal candidate genes and pathways for residual feed intake. Front. Genet. 2014, 5, 307. [Google Scholar] [CrossRef] [Green Version]
- Lkhagvadorj, S.; Qu, L.; Cai, W.G.; Couture, O.P.; Barb, C.R.; Hausman, G.J.; Nettleton, D.; Anderson, L.L.; Dekkers, J.C.M.; Tuggle, C.K. Gene expression profiling of the short-term adaptive response to acute caloric restriction in liver and adipose tissues of pigs differing in feed efficiency. Am. J. Physiol. 2010, 298, R494–R507. [Google Scholar] [CrossRef]
- Vincent, A.; Louveau, I.; Gondret, F.; Trefeu, C.; Gilbert, H.; Lefaucheur, L. Divergent selection for residual feed intake affects the transcriptomic and proteomic profiles of pig skeletal muscle. J. Anim. Sci. 2015, 93, 2745–2758. [Google Scholar] [CrossRef]
- Gondret, F.; Vincent, A.; Houee-Bigot, M.; Siegel, A.; Lagarrigue, S.; Causeur, D.; Gilbert, H.; Louveau, I. A transcriptome multi-tissue analysis identifies biological pathways and genes associated with variations in feed efficiency of growing pigs. BMC Genom. 2017, 18, 244. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.; Hou, Y.; Wu, H.; Miao, Y.; Li, X.; Cao, J.; Brameld, J.M.; Parr, T.; Zhao, S. Transcriptome analysis of mRNA and miRNA in skeletal muscle indicates an important network for differential Residual Feed Intake in pigs. Sci. Rep. 2015, 5, 11953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.X.; Hou, Y.; Liu, F.; Liu, A.; Jing, L.; Zhao, C.Z.; Luan, Y.; Miao, Y.X.; Zhao, S.H.; Li, X.Y. Transcriptome Analysis Reveals that Vitamin A Metabolism in the Liver Affects Feed Efficiency in Pigs. G3-Genes Genom Genet. 2016, 6, 3615–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louveau, I.; Vincent, A.; Tacher, S.; Gilbert, H.; Gondret, F. Increased expressions of genes and proteins involved in mitochondrial oxidation and antioxidant pathway in adipose tissue of pigs selected for a low residual feed intake. J. Anim. Sci. 2016, 94, 5042–5054. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.B.; Nguyen, Y.T.; Nettleton, D.; Dekkers, J.C.M.; Tuggle, C.K. Post-weaning blood transcriptomic differences between Yorkshire pigs divergently selected for residual feed intake. BMC Genom. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Ramayo-Caldas, Y.; Ballester, M.; Sanchez, J.P.; Gonzalez-Rodriguez, O.; Revilla, M.; Reyer, H.; Wimmers, K.; Torrallardona, D.; Quintanilla, R. Integrative approach using liver and duodenum RNA-Seq data identifies candidate genes and pathways associated with feed efficiency in pigs. Sci. Rep. 2018, 8, 558. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Wang, Y.; Yang, T.; Xing, K.; Ao, H.; Chen, S.; Zhang, F.; Zhao, X.; Liu, J.; Wang, C. Differentially expressed genes in the caecal and colonic mucosa of Landrace finishing pigs with high and low food conversion ratios. Sci. Rep. 2017, 7, 14886. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 21 May 2010).
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Basel, Switzerland, 2016. [Google Scholar]
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C T method. Nat. Protoc. 2008, 3, 1101. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Metzler-Zebell, B.U.; Lawlor, P.G.; Magowan, E.; McCormack, U.M.; Curiao, T.; Hollmann, M.; Ertl, R.; Aschenbach, J.R.; Zebell, Q. Finishing pigs that are divergent in feed efficiency show small differences in intestinal functionality and structure. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Vigors, S.; Sweeney, T.; O’Shea, C.J.; Kelly, A.K.; O’Doherty, J.V. Pigs that are divergent in feed efficiency, differ in intestinal enzyme and nutrient transporter gene expression, nutrient digestibility and microbial activity. Animal 2016, 10, 1848–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- Roncero, I.; Alvarez, E.; Chowen, J.; Sanz, C.; R¢bano, A.; V¨¢zquez, P.; Bl¢zquez, E. Expression of glucose transporter isoform GLUT-2 and glucokinase genes in human brain. J. Neurochem. 2004, 88, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Yang, X.; Gu, Y.; Tao, X.; Zhong, Z.; Gong, J.; Chen, X.; Lv, X. Developmental changes in the expression of the GLUT2 and GLUT4 genes in the longissimus dorsi muscle of Yorkshire and Tibetan pigs. Genet. Mol. Res. 2015, 14, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Bedford, A.; Huynh, E.; Fu, M.; Zhu, C.; Wey, D.; de Lange, C.; Li, J. Growth performance of early-weaned pigs is enhanced by feeding epidermal growth factor-expressing Lactococcus lactis fermentation product. J. Biotechnol. 2014, 173, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhu, F.; Yang, H.; Li, J.; Li, Y.; Ding, X.; Xiong, X.; Yin, Y. Effects of dietary supplementation with epidermal growth factor on nutrient digestibility, intestinal development and expression of nutrient transporters in early-weaned piglets. J. Anim. Physiol. 2019, 103, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.; Aranias, T.; Viel, T.; Chateau, D.; Le Gall, M.; Waligora-Dupriet, A.; Melchior, C.; Rouxel, O.; Kapel, N.; Gourcerol, G.; et al. Intestinal invalidation of the glucose transporter GLUT2 delays tissue distribution of glucose and reveals an unexpected role in gut homeostasis. Mol. Metab. 2017, 6, 61–72. [Google Scholar] [CrossRef]
- Dai, L.; Hu, W.; Xia, L.; Xia, M.; Yang, Q. Transmissible Gastroenteritis Virus Infection Enhances SGLT1 and GLUT2 Expression to Increase Glucose Uptake. PLoS ONE 2016, 11, e0165585. [Google Scholar] [CrossRef]
- Guillam, M.; Hmmler, E.; Schaerer, E.; Yeh, J.; Birnbaum, M.; Beermann, F.; Schmidt, A.; Driaz, N.; Thorens, B. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat. Genet. 1997, 17, 327–330. [Google Scholar] [CrossRef]
- Guillam, M.; Dupraz, P.; Thorens, B. Glucose uptake, utilization, and signaling in GLUT2-null islets. Diabetes 2000, 49, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Steinert, R.E.; Beglinger, C.; Langhans, W. Intestinal GLP-1 and satiation—From man to rodents and back. Int. J. Obes. 2016, 40. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Hill, M.; Asa, S.L.; Brubaker, P.L.; Drucker, D.J. Intestinal growth-promoting properties of glucagon-like peptide-2 in mice. Am. J. Physiol. 1997, 273, E77–E84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munroe, D.G.; Gupta, A.K.; Kooshesh, F.; Vyas, T.B.; Rizkalla, G.; Wang, H.; Demchyshyn, L.; Yang, Z.-J.; Kamboj, R.K.; Chen, H. Prototypic G protein-coupled receptor for the intestinotrophic factor glucagon-like peptide 2. Proc. Natl. Acad. Sci. USA 1999, 96, 1569–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrin, D.; Stoll, B.; Jiang, R.; Petersen, Y.; Elnif, J.; Buddington, R.; Schmidt, M.; Holst, J.; Hartmann, B.; Sangild, P. GLP-2 stimulates intestinal growth in premature TPN-fed pigs by suppressing proteolysis and apoptosis. Am. J. Physiol. 2000, 279, G1249–G1256. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Yu, D.; Schwartz, M.Z. Glucagonlike peptide-2 enhances small intestinal absorptive function and mucosal mass in vivo. J. Pediatric Surg. 1999, 34, 18–21. [Google Scholar] [CrossRef]
- Reyer, H.; Oster, M.; Magowan, E.; Murani, E.; Sauerwein, H.; Dannenberger, D.; Kuhla, B.; Ponsuksili, S.; Wimmers, K. Feed-efficient pigs exhibit molecular patterns allowing a timely circulation of hormones and nutrients. Physiol Genom. 2018, 50, 726–734. [Google Scholar] [CrossRef]
- Oczkowicz, M.; Ropka-Molik, K.; Tyra, M. Analysis of the associations between polymorphisms in GNAS complex locus and growth, carcass and meat quality traits in pigs. Mol. Biol. Rep. 2013, 40, 6419–6427. [Google Scholar] [CrossRef]
- Tyra, M.; Ropka-Molik, K.; Piórkowska, K.; Oczkowicz, M.; Szyndler-Nędza, M.; Małopolska, M. Association of Ghrelin Gene Polymorphisms with Fattening Traits and Feed Intake in Pig: A Preliminary Study. Animals 2019, 9, 410. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | HE (n = 6) | LE (n = 6) | p-value |
|---|---|---|---|
| SW (kg) | 30.87 ± 1.54 | 30.13 ± 0.76 | 3.30 × 10−1 |
| EW (kg) | 100.23 ± 0.62 | 99.37 ± 1.17 | 1.48 × 10−1 |
| TD (day) | 82.17 ± 5.27 | 90.83 ± 3.49 | 8.85 × 10−3 |
| ADG (kg/day) | 0.85 ± 0.05 | 0.77 ± 0.03 | 1.14 × 10−2 |
| DFI (kg) | 1.83 ± 0.15 | 2.03 ± 0.13 | 3.70 × 10−2 |
| FCR (kg/kg) | 2.19 ± 0.08 | 2.68 ± 0.05 | 8.91 × 10−7 |
| AMBW (kg) | 23.04 ± 0.24 | 22.83 ± 0.21 | 1.37 × 10−1 |
| RFI (kg/day) | −0.18 ± 0.08 | 0.14 ± 0.09 | 7.19 × 10−5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Li, S.; Wu, J.; Ding, R.; Quan, J.; Zheng, E.; Yang, J.; Wu, Z. A Transcriptome Analysis Identifies Biological Pathways and Candidate Genes for Feed Efficiency in DLY Pigs. Genes 2019, 10, 725. https://doi.org/10.3390/genes10090725
Wang X, Li S, Wu J, Ding R, Quan J, Zheng E, Yang J, Wu Z. A Transcriptome Analysis Identifies Biological Pathways and Candidate Genes for Feed Efficiency in DLY Pigs. Genes. 2019; 10(9):725. https://doi.org/10.3390/genes10090725
Chicago/Turabian StyleWang, Xingwang, Shaoyun Li, Jie Wu, Rongrong Ding, Jianping Quan, Enqin Zheng, Jie Yang, and Zhenfang Wu. 2019. "A Transcriptome Analysis Identifies Biological Pathways and Candidate Genes for Feed Efficiency in DLY Pigs" Genes 10, no. 9: 725. https://doi.org/10.3390/genes10090725