Characterization of the Complete Mitochondrial Genome of Harpalus sinicus and Its Implications for Phylogenetic Analyses


Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Sequencing and Genome Assembly
2.2. Genome Sequencing, Assembly, and Annotation
2.3. Sequence Analysis
2.4. Phylogenetic Analysis
3. Results and Discussion
3.1. The Organization and Nucleotide Composition of the Mitogenome of H. sinicus
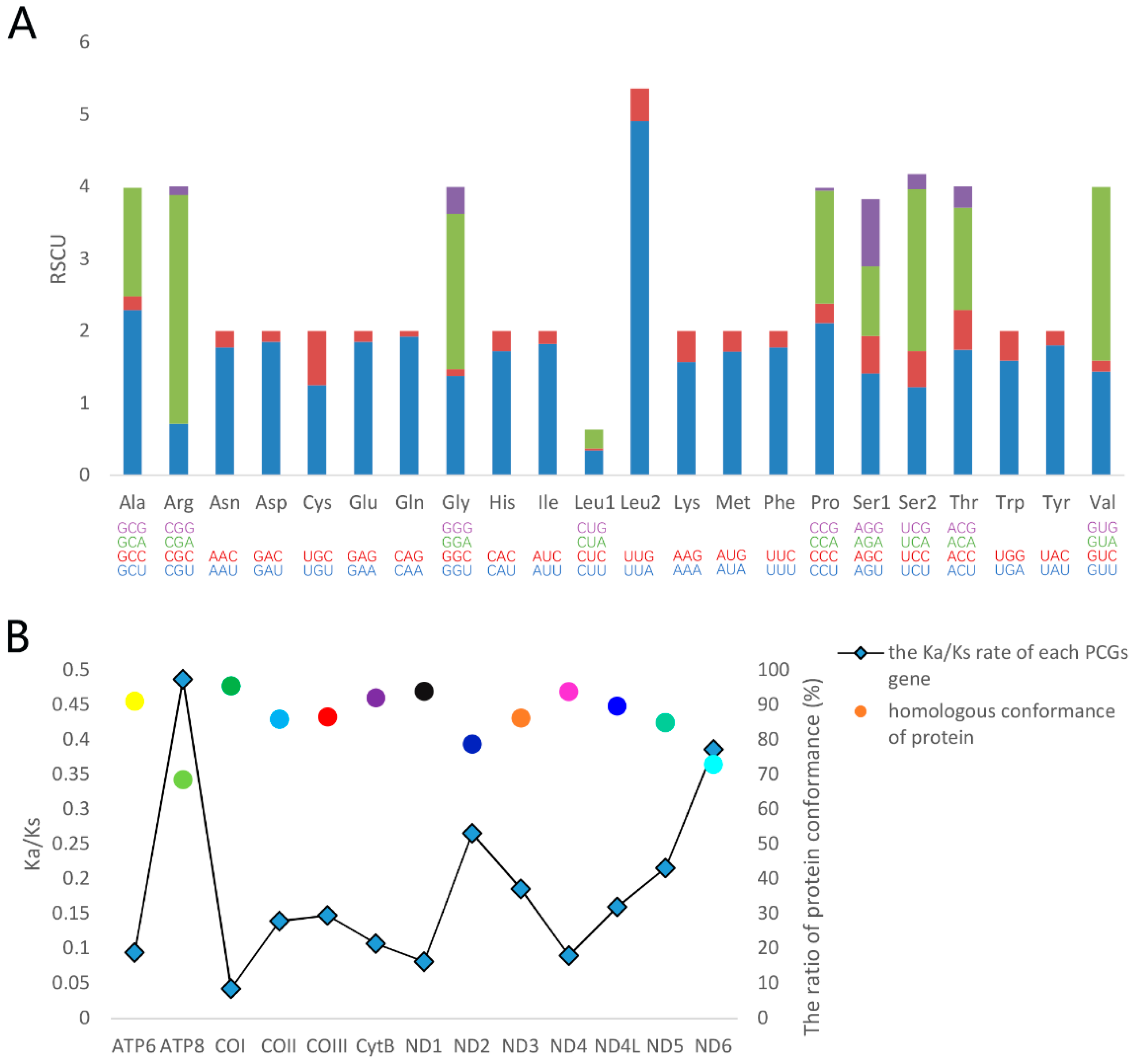
3.2. Protein-Coding Genes
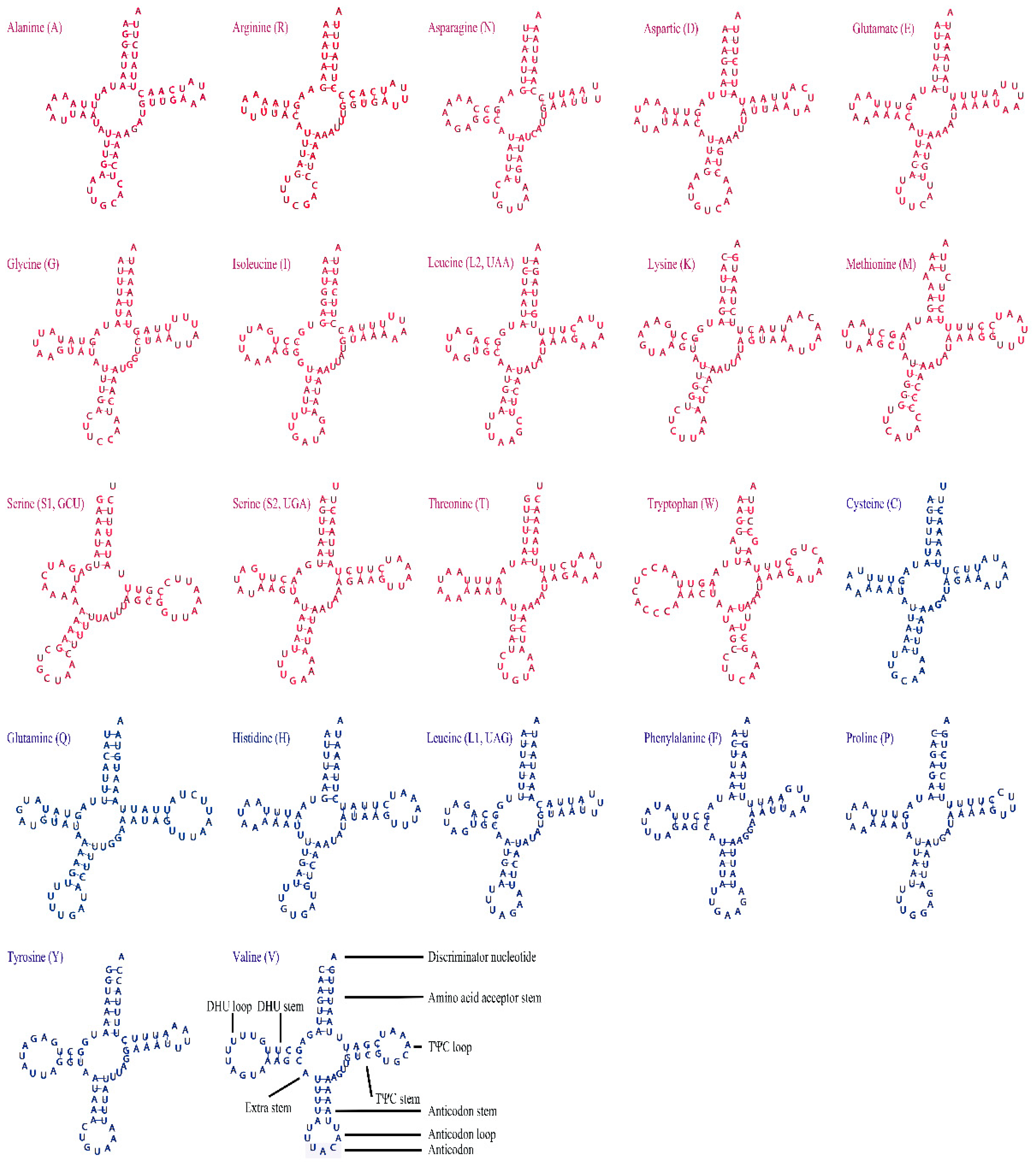
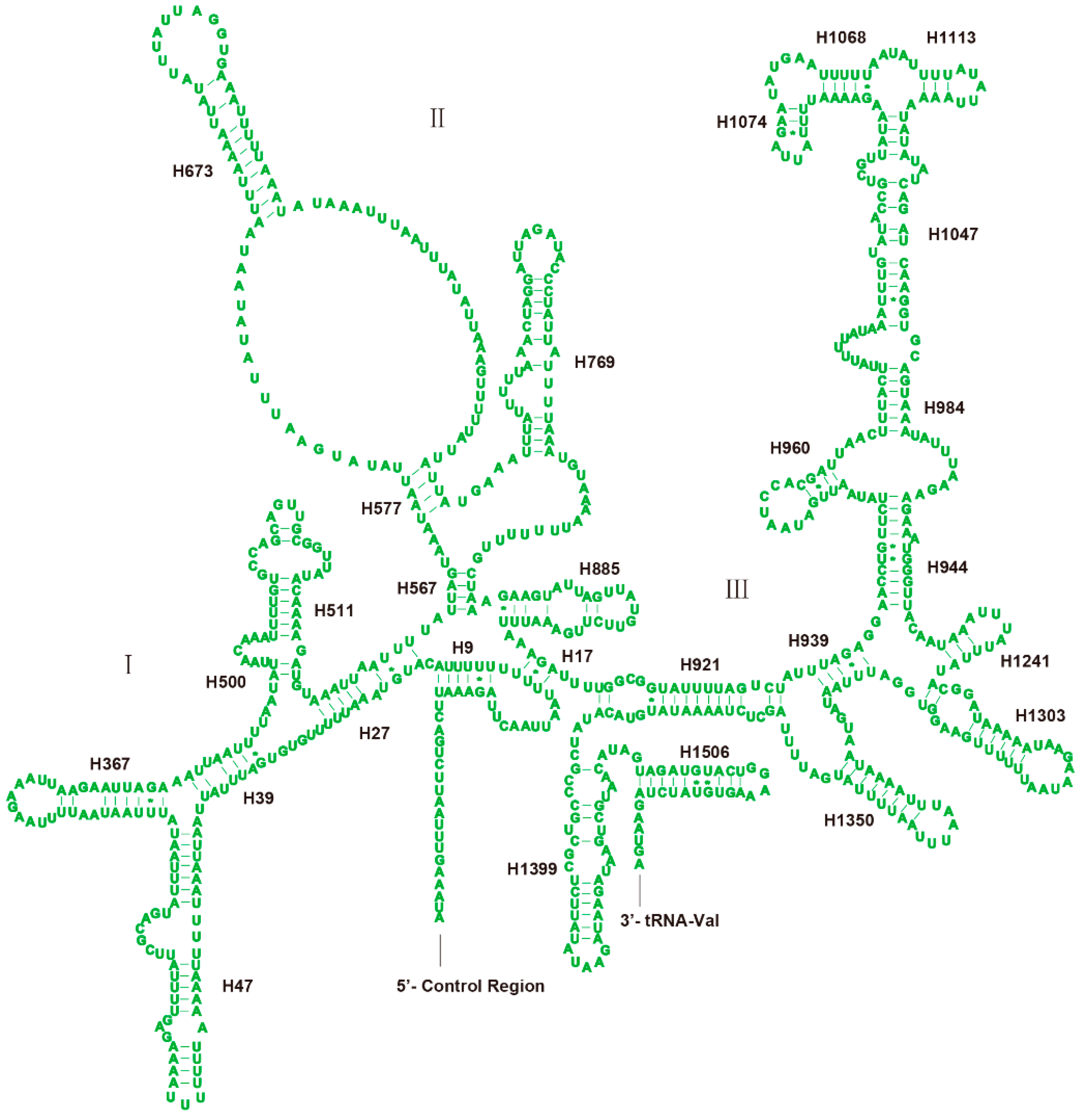
3.3. Transfer RNAs
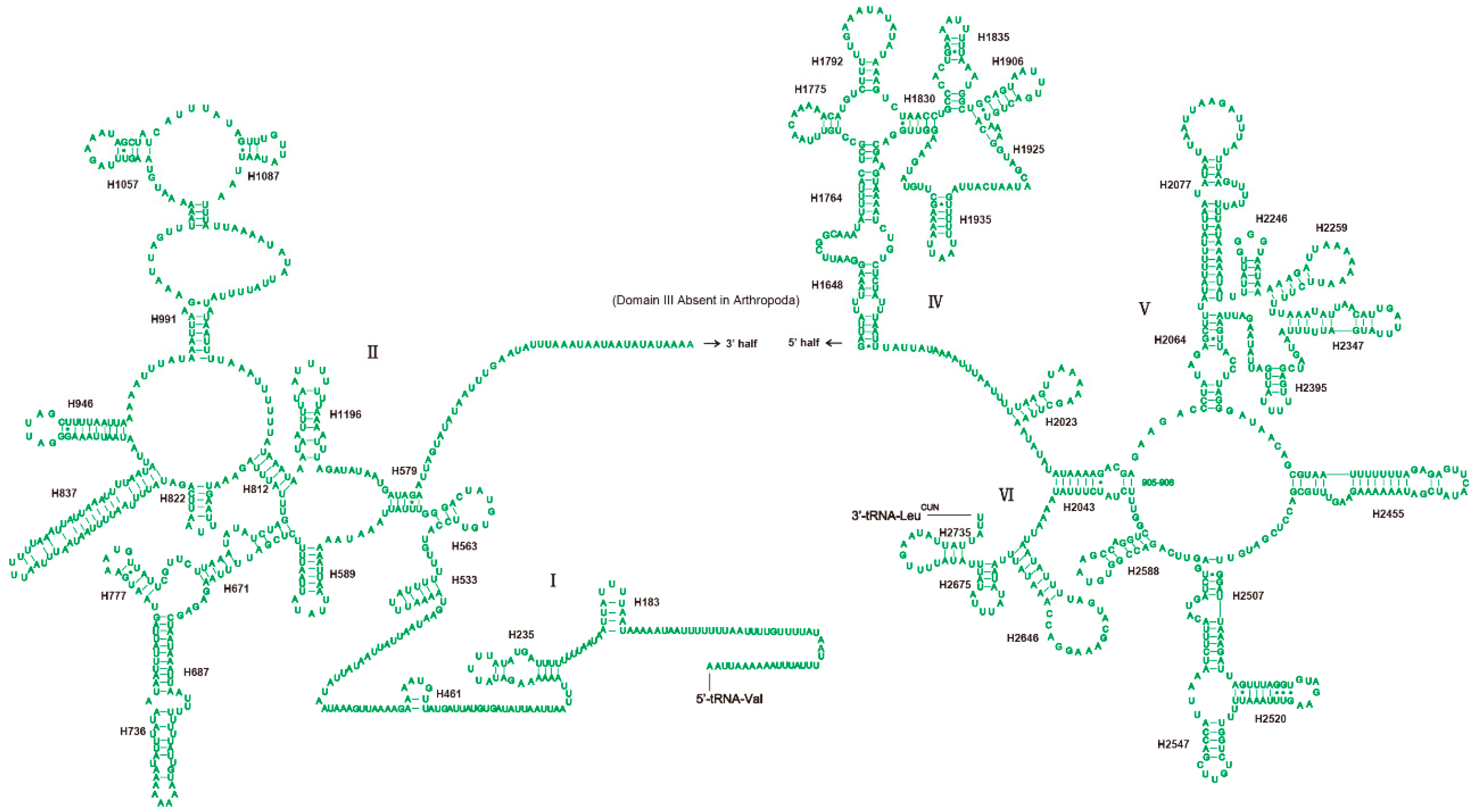
3.4. Ribosomal RNAs
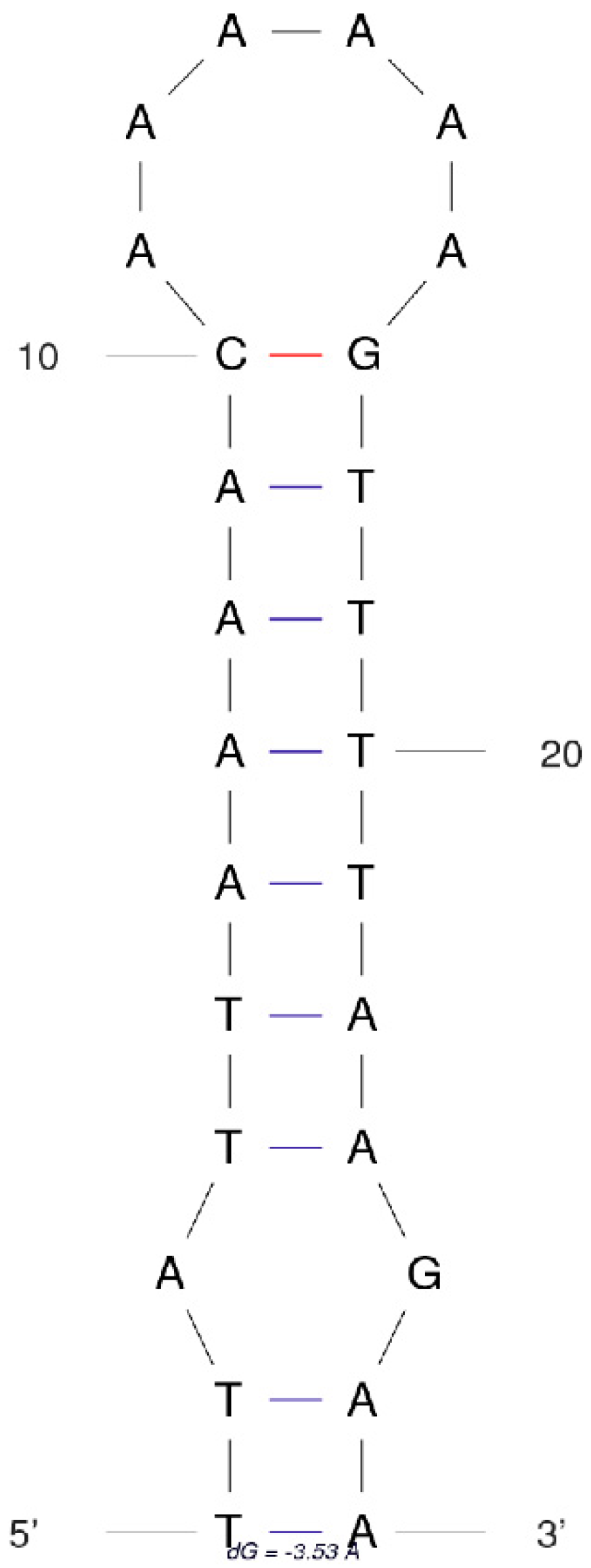
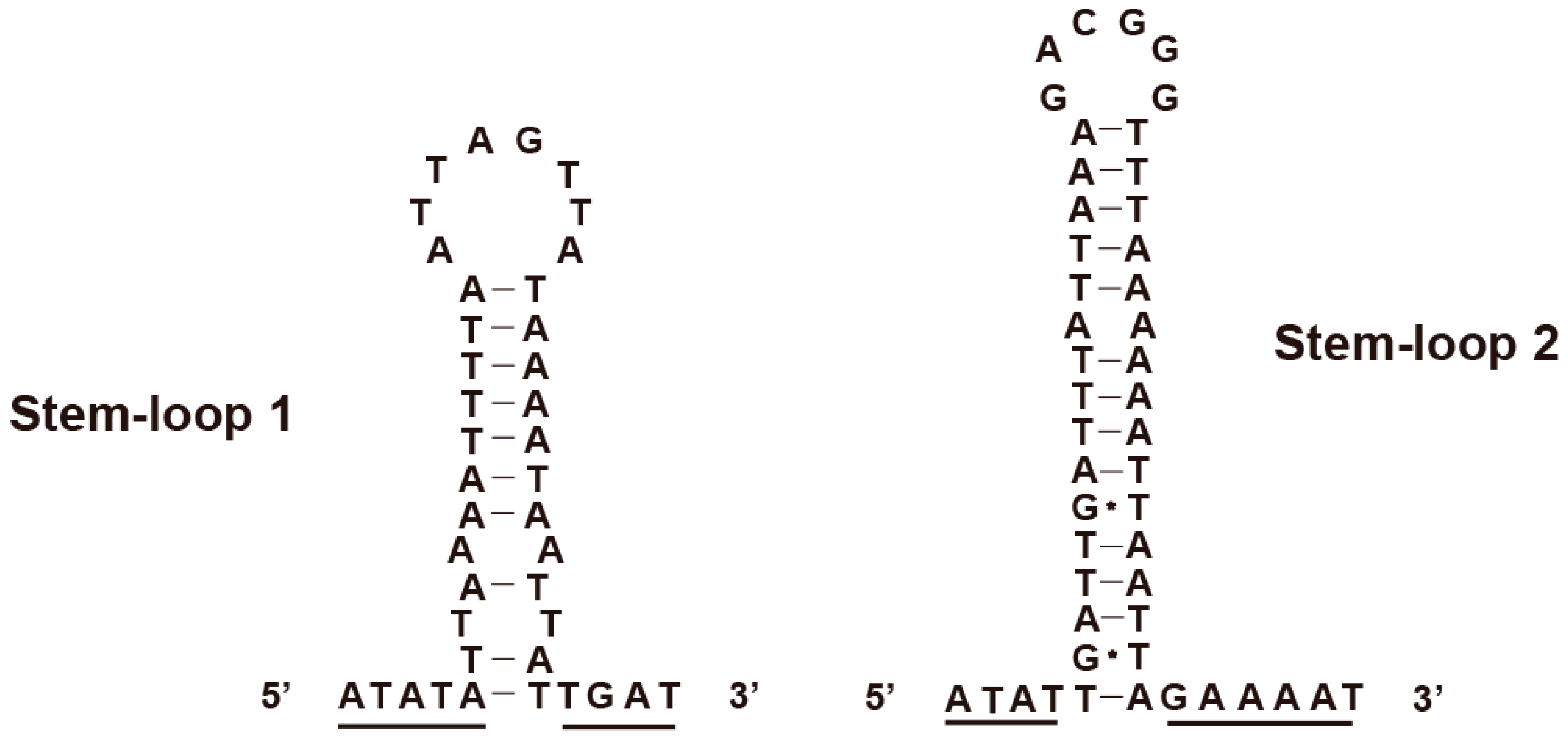
3.5. Control Region
3.6. Phylogenetic Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gabaldon, T.; Huynen, M.A. Reconstruction of the proto-mitochondrial metabolism. Science 2003, 301, 609. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Xu, G.; Gu, B.; Shi, Y.; Mzuka, H.L.; Shen, H. The Complete Mitochondrial Genome Sequences of the Philomycus bilineatus (Stylommatophora: Philomycidae) and Phylogenetic Analysis. Genes 2019, 10, 198. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.P.; Danforth, B.N. How do insect nuclear and mitochondrial gene substitution patterns differ? Insights from Bayesian analyses of combined datasets. Mol. Phylogenet. Evol. 2004, 30, 686–702. [Google Scholar] [CrossRef]
- Ahmad, A.A.; Yang, X.; Zhang, T.; Wang, C.; Zhou, C.; Yan, X.; Hassan, M.; Ikram, M.; Hu, M. Characterization of the Complete Mitochondrial Genome of Ostertagia trifurcata of Small Ruminants and its Phylogenetic Associations for the Trichostrongyloidea Superfamily. Genes 2019, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Gao, C.; Cui, Y.; Xie, Q.; Bu, W. The complete mitochondrial genome of the stalk-eyed bug Chauliops fallax Scott, and the monophyly of Malcidae (Hemiptera: Heteroptera). PLoS ONE 2013, 8, e55381. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Lin, A.; Zhao, X. Insight into higher-level phylogeny of Neuropterida: Evidence from secondary structures of mitochondrial rRNA genes and mitogenomic data. PLoS ONE 2018, 13, e0191826. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Yang, M.F.; Dai, R.H.; Li, H.; Wang, X.Y. Characterization and phylogenetic implications of the complete mitochondrial genome of Idiocerinae (Hemiptera: Cicadellidae). Int. J. Biol. Macromol. 2018, 120, 2366–2372. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.T.; Wu, T.L.; Wu, X.L.; Deng, W. Description of Natural Enemies of Pasture Pest-Car-abdae Insects. Plant Dis. Pests 2010, 1, 41–44. [Google Scholar] [CrossRef]
- Zhao, T.G.; Wu, T.L.; Mo, X.L.; Wu, X.J. Predatory Enemy of Harpalus griseus (panzer) against Pasture Pests. Plant Dis. Pests 2010, 1, 60–62. [Google Scholar] [CrossRef]
- Zheng, L.Y.; Gui, H. Insect Classification; Nanjing Normal University Press: Nanjing, China, 1999. [Google Scholar]
- Kromp, B. Carabid beetles in sustainable agriculture: A review on pest control efficacy, cultivation impacts and enhancement. Agric. Ecosyst. Environ. 1999, 74, 187–228. [Google Scholar] [CrossRef]
- Sun, L.R.; Qi, Y.J.; Tian, X.X. Analysis of mitochondrial genome of Scolopendra subspinipes dehaani. Tianjin J. Tradit. Chin. Med. 2018, 35, 225–229. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2013. [Google Scholar]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinform. (Oxf. Engl.) 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, J.J.; Li, W.H. Complete Mitochondrial Genome of Suwallia teleckojensis (Plecoptera: Chloroperlidae) and Implications for the Higher Phylogeny of Stoneflies. Int. J. Mol. Sci. 2018, 680, 107. [Google Scholar] [CrossRef]
- Li, T.; Yang, J.; Li, Y.; Cui, Y.; Xie, Q.; Bu, W.; Hillis, D.M. A Mitochondrial Genome of Rhyparochromidae (Hemiptera: Heteroptera) and a Comparative Analysis of Related Mitochondrial Genomes. Sci. Rep. 2016, 6, 35175. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Li, W.; Jakovlić, I.; Zou, H.; Zhang, J.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. bioRxiv 2018. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Characterization and Phylogenetic Implications of the Complete Mitochondrial Genome of Syrphidae. Genes 2019, 10, 563. [Google Scholar] [CrossRef]
- Bao, L.; Zhang, Y.; Gu, X.; Gao, Y.; Yu, Y. The complete mitochondrial genome of Eterusia aedea (Lepidoptera, Zygaenidae) and comparison with other zygaenid moths. Genomics 2018. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H.; Krishnan, N.M.; Rao, B.J. Possible multiple origins of replication in primate mitochondria: Alternative role of tRNA sequences. J. Theor. Biol. 2006, 241, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H.; Krishnan, N.M.; Rao, B.J. Mitochondrial tRNA sequences as unusual replication origins: Pathogenic implications for Homo sapiens. J. Theor. Biol. 2006, 243, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H. Hybridization between mitochondrial heavy strand tDNA and expressed light strand tRNA modulates the function of heavy strand tDNA as light strand replication origin. J. Mol. Biol. 2008, 379, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H. Avoidance of antisense, antiterminator tRNA anticodons in vertebrate mitochondria. Bio Syst. 2010, 101, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H.; Labra, A. The relation between hairpin formation by mitochondrial WANCY tRNAs and the occurrence of the light strand replication origin in Lepidosauria. Gene 2014, 542, 248–257. [Google Scholar] [CrossRef]
- Seligmann, H.; Krishnan, N.M. Mitochondrial replication origin stability and propensity of adjacent tRNA genes to form putative replication origins increase developmental stability in lizards. J. Exp. Zool. Part B Mol. Dev. Evol. 2006, 306, 433–449. [Google Scholar] [CrossRef]
- Fuste, J.M.; Wanrooij, S.; Jemt, E.; Granycome, C.E.; Cluett, T.J.; Shi, Y.; Atanassova, N.; Holt, I.J.; Gustafsson, C.M.; Falkenberg, M. Mitochondrial RNA polymerase is needed for activation of the origin of light-strand DNA replication. Mol. Cell 2010, 37, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H. Mitochondrial tRNAs as light strand replication origins: Similarity between anticodon loops and the loop of the light strand replication origin predicts initiation of DNA replication. Biosystems 2010, 99, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Hua, J.; Wright, A.M.; Cui, Y.; Xie, Q.; Bu, W.; Hillis, D.M. Long-branch attraction and the phylogeny of true water bugs (Hemiptera: Nepomorpha) as estimated from mitochondrial genomes. BMC Evol. Biol. 2014, 14, 99. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liao, M.; Yang, M.; Xiong, C.; Jin, X.; Chen, Z.; Huang, W. Characterization of the mitochondrial genomes of three species in the ectomycorrhizal genus Cantharellus and phylogeny of Agaricomycetes. Int. J. Biol. Macromol. 2018, 118, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.R.; Wu, W.; Goodman, M.; Grossman, L.I. Evolution of nuclear- and mitochondrial-encoded subunit interaction in cytochrome c oxidase. Mol. Biol. Evol. 2001, 18, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.; Ratnasingham, S.; deWaard, J.R. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc. Biol. Sci. 2003, 270, S96–S99. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Jiang, L.Y.; Qiao, G.X. Hemipteran mitochondrial genomes: Features, structures and implications for phylogeny. Int. J. Mol. Sci. 2015, 16, 12382–12404. [Google Scholar] [CrossRef] [PubMed]
- Gautheret, D.; Konings, D.; Gutell, R.R.G. U base pairing motifs in ribosomal RNA. RNA 1995, 1, 807–814. [Google Scholar] [PubMed]
- Yokobori, S.I.; Paabo, S. tRNA editing in metazoans. Nature 1995, 377, 490. [Google Scholar] [CrossRef]
- He, B.; Su, T.; Niu, Z.; Zhou, Z.; Gu, Z.; Huang, D. Characterization of mitochondrial genomes of three Andrena bees (Apoidea: Andrenidae) and insights into the phylogenetics. Int. J. Biol. Macromol. 2019, 127, 118–125. [Google Scholar] [CrossRef]
- Zhang, D.X.; Godfrey, M.H. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 1997, 25, 99–120. [Google Scholar] [CrossRef]
- Zhang, D.X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.J.; Zhu, W.C.; Rong, X.; Zhang, Y.K.; Ding, X.L.; Liu, J.; Chen, D.S.; Du, Y.; Hong, X.Y. The complete mitochondrial genomes of two rice planthoppers, Nilaparvata lugens and Laodelphax striatellus: Conserved genome rearrangement in Delphacidae and discovery of new characteristics of atp8 and tRNA genes. BMC Genom. 2013, 14, 417. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, A.; Vogler, A.P. The mitogenome phylogeny of Adephaga (Coleoptera). Mol. Phylogenet. Evol. 2017, 114, 166–174. [Google Scholar] [CrossRef] [PubMed]
- David, M.; Michael, D.B.; Ober, K.A. Phylogeny of Carabid beetles as inferred from 18S ribosomal DNA (Coleoptera: Carabidae). Syst. Entomol. 1999, 24, 103–138. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Strand | Location | Size (bp) | Start Condon | Stop Codon | Anticodon | Spacer (+)/Overlap (−) |
|---|---|---|---|---|---|---|---|
| tRNA-Ile | J | 1–68 | 68 | GAT | 0 | ||
| tRNA-Gln | N | 66–137 | 72 | TTG | −3 | ||
| tRNA-Met | J | 143–211 | 69 | CAT | 5 | ||
| ND2 | J | 212–1231 | 1020 | ATA | TAA | 0 | |
| tRNA-Trp | J | 1237–1309 | 73 | TCA | 5 | ||
| tRNA-Cys | N | 1338–1402 | 65 | GCA | 28 | ||
| tRNA-Tyr | N | 1409–1476 | 68 | GTA | 6 | ||
| COI | J | 1469–3010 | 1542 | ATT | TAA | −8 | |
| tRNA-Leu | J | 3013–3077 | 65 | TAA | 2 | ||
| COII | J | 3079–3762 | 684 | ATG | TAA | 1 | |
| tRNA-Lys | J | 3773–3843 | 71 | CTT | 10 | ||
| tRNA-Asp | J | 3844–3911 | 68 | GTT | 0 | ||
| ATP8 | J | 3912–4073 | 162 | ATC | TAA | 0 | |
| ATP6 | J | 4067–4744 | 678 | ATG | TAA | −7 | |
| COIII | J | 4751–5539 | 789 | ATG | TAA | 6 | |
| tRNA-Gly | J | 5541–5606 | 66 | TCC | 1 | ||
| ND3 | J | 5607–5960 | 354 | ATT | TAG | 0 | |
| tRNA-Ala | J | 5959–6024 | 66 | TGC | −2 | ||
| tRNA-Arg | J | 6025–6090 | 66 | TCG | 0 | ||
| tRNA-Asn | J | 6093–6157 | 65 | GTT | 2 | ||
| tRNA-Ser | J | 6158–6224 | 67 | GCT | 0 | ||
| tRNA-Glu | J | 6226–6292 | 67 | TCC | 1 | ||
| tRNA-Phe | N | 6291–6357 | 97 | TGG | −2 | ||
| ND5 | N | 6359–8086 | 1728 | ATT | A- | 1 | |
| tRNA-His | N | 8087–8155 | 69 | GTG | 0 | ||
| ND4 | N | 8155–9495 | 1341 | ATG | TAA | −1 | |
| ND4L | N | 9489–9785 | 297 | ATT | TAA | −7 | |
| tRNA-Thr | J | 9788–9853 | 66 | TGT | 2 | ||
| tRNA-Pro | N | 9856–9919 | 64 | TGG | 2 | ||
| ND6 | J | 9921–10,445 | 525 | ATT | TAA | 1 | |
| CytB | J | 10,446–11,585 | 1140 | ATG | TAG | 0 | |
| tRNA-Ser | J | 11,584–11,651 | 68 | TGA | −2 | ||
| ND1 | N | 11,669–12,619 | 951 | TTG | TAG | 17 | |
| tRNA-Leu | N | 12,621–12,684 | 64 | TAG | 1 | ||
| rrnL | N | 12,685–14,010 | 1326 | 0 | |||
| tRNA-Val | N | 14,013–14,085 | 73 | TAC | 2 | ||
| rrnS | N | 14,086–14,873 | 788 | 0 | |||
| Control region | 14,874–16,524 | 1651 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Tan, W.; Zhang, H.; Jiang, W.; Gao, H.; Wang, W.; Liu, Y.; Wang, Y.; Tian, X. Characterization of the Complete Mitochondrial Genome of Harpalus sinicus and Its Implications for Phylogenetic Analyses. Genes 2019, 10, 724. https://doi.org/10.3390/genes10090724
Yu X, Tan W, Zhang H, Jiang W, Gao H, Wang W, Liu Y, Wang Y, Tian X. Characterization of the Complete Mitochondrial Genome of Harpalus sinicus and Its Implications for Phylogenetic Analyses. Genes. 2019; 10(9):724. https://doi.org/10.3390/genes10090724
Chicago/Turabian StyleYu, Xiaolei, Wei Tan, Huanyu Zhang, Weiling Jiang, Han Gao, Wenxiu Wang, Yuxia Liu, Yu Wang, and Xiaoxuan Tian. 2019. "Characterization of the Complete Mitochondrial Genome of Harpalus sinicus and Its Implications for Phylogenetic Analyses" Genes 10, no. 9: 724. https://doi.org/10.3390/genes10090724