Identifying Interaction Clusters for MiRNA and MRNA Pairs in TCGA Network

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Significant mRNA–miRNA Pairs Selection for Input Data
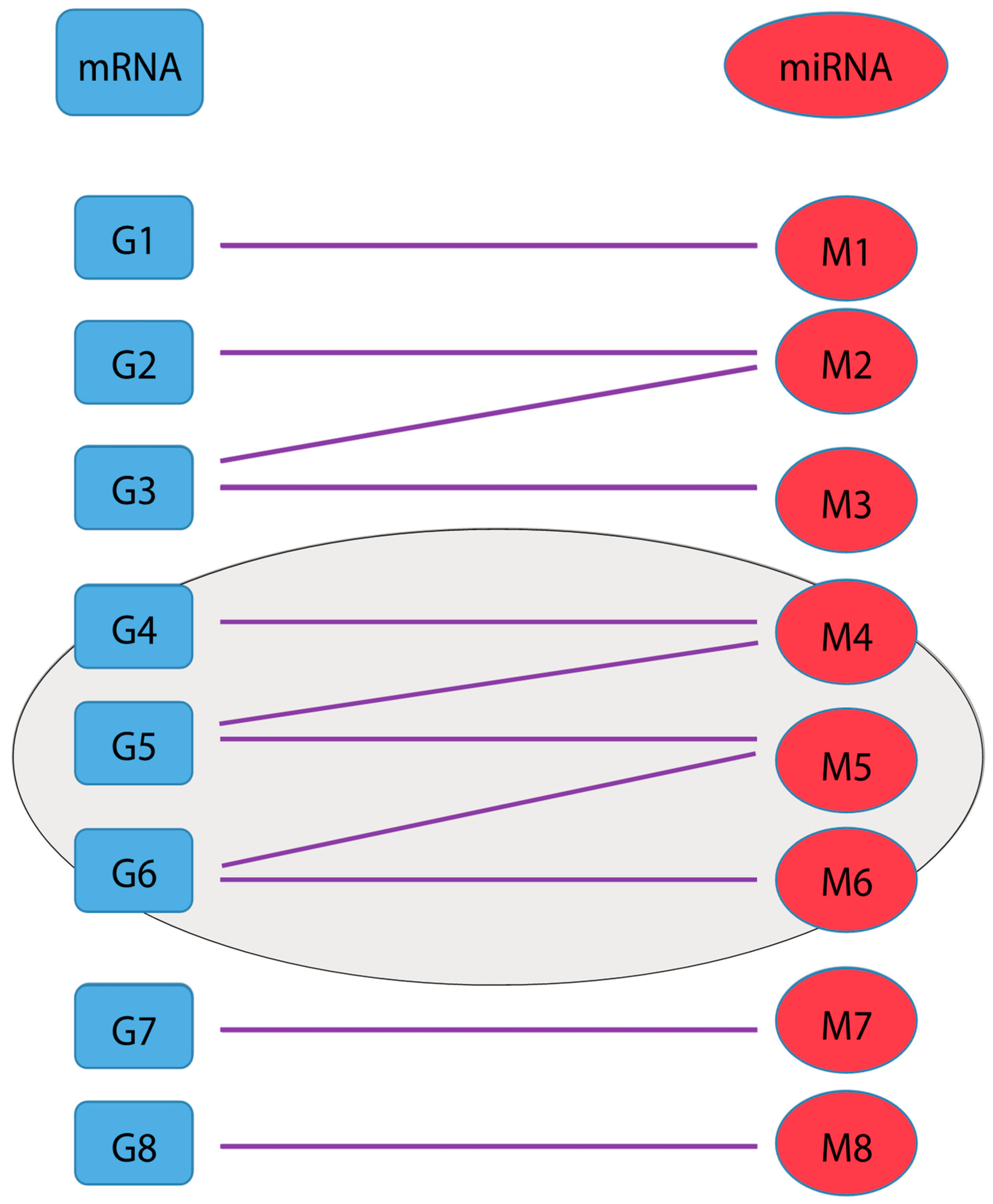
2.2. Cluster Identification and Scoring Algorithm
2.3. Statistical Analysis
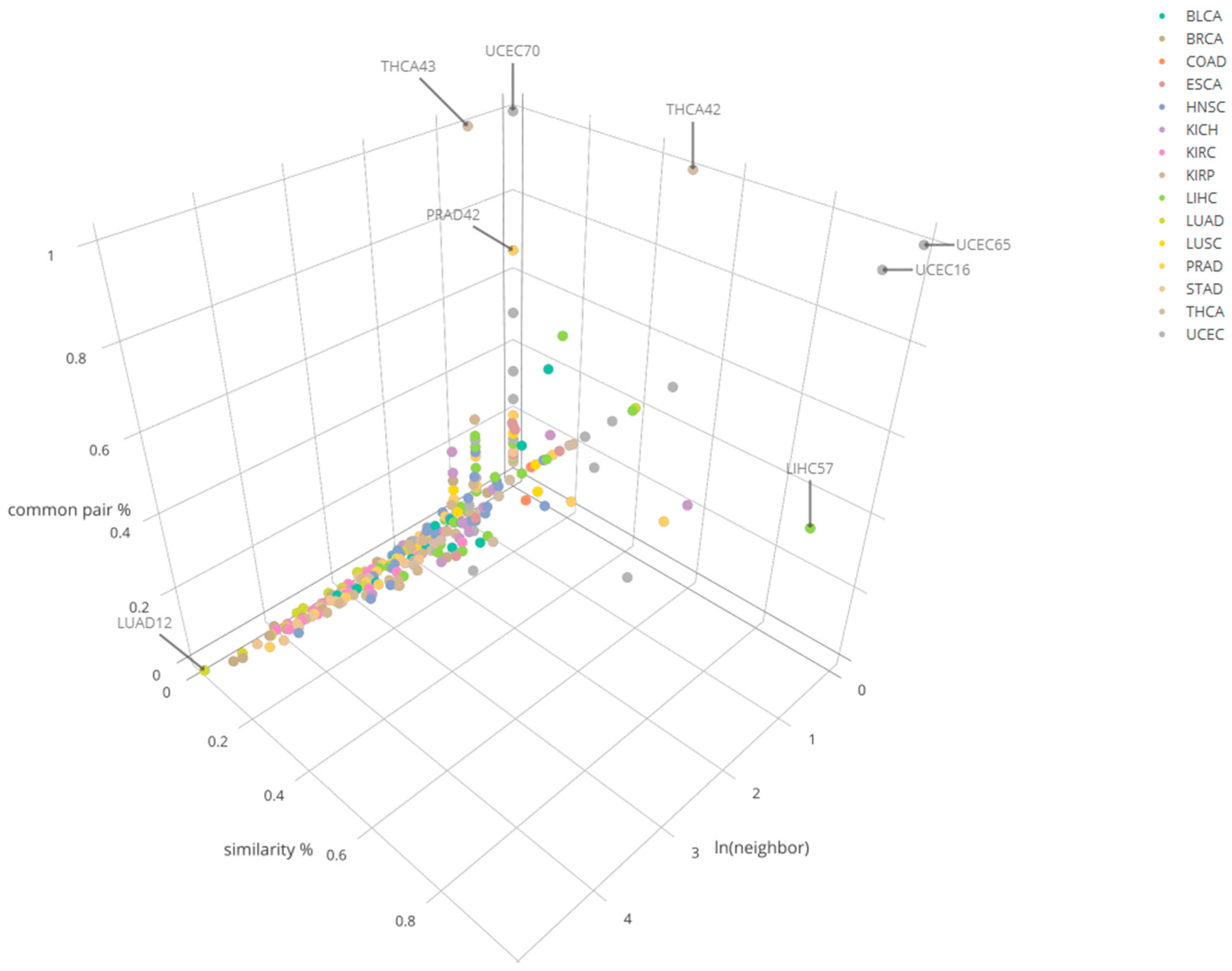
2.4. Determination of Shared Clusters Across Tumor Types and/or Subtypes
- Identify corresponding vertices (genes and miRNAs) and edges (their interactions) that connect their vertices in clusters between two different cancers (A and B);
- Construct matrices to store vertices and edges;
- Calculate the shortest “distance” as the number of edges between any two vertices for each cluster in cancer A and B, respectively;
- Determine whether two clusters match based on their matching percentage (defined as the ratio of the number of corresponding vertex pairs with equal distance out of total matched vertex pairs).
2.5. Check the Overlap Between miRNAs Reported in LIHC and Differentially Expressed miRNAs from Studies of Human and Rat with Nonalcoholic Fatty Liver Disease
3. Results
3.1. Inversely Correlated miRNA and mRNA Pairs with Opposite Fold Change
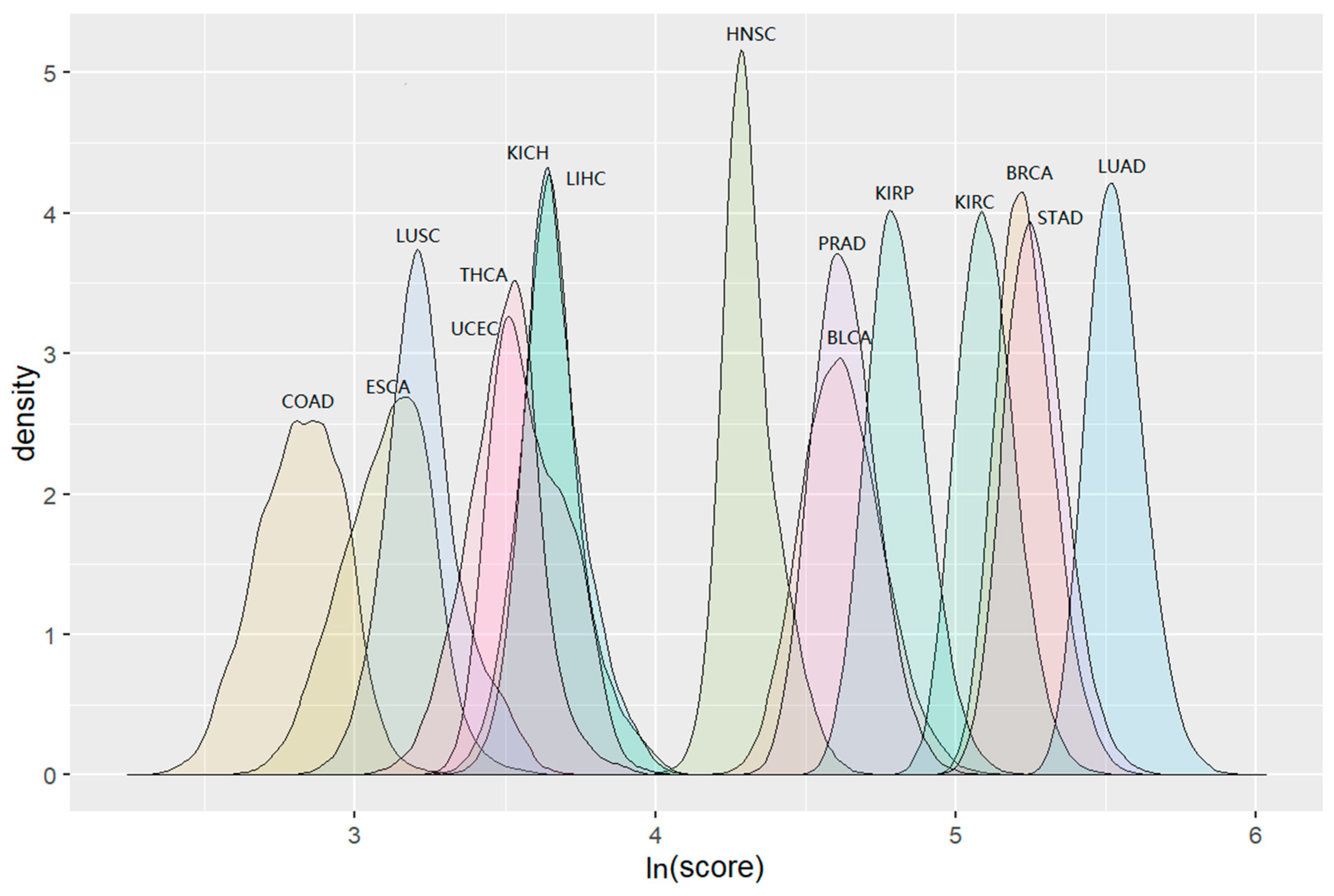
3.2. Cluster Detection Results
3.3. Cross-Cancer Comparison Results
3.4. Investigation of miRNAs and Their Targets Overlap Between Lists in LIHC and the Ones Reported from the Study in Human with Nonalcoholic Fatty Liver Disease
3.5. Investigation of miRNA Overlap Between Differentially Expressed miRNA List in Rat with Nonalcoholic Fatty Liver Disease and the Ones in LIHC
3.6. Cluster Functional Analysis for LIHC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TCGA | The Cancer Genome Atlas |
| LIHC | Liver hepatocellular carcinoma |
| KIRC | Kidney renal clear cell carcinoma |
| KIRP | Kidney renal papillary cell carcinoma |
| LUAD | Lung adenocarcinoma |
| BLCA | Bladder urothelial carcinoma |
| BRCA | Breast invasive carcinoma |
| COAD | Colon adenocarcinoma |
| ESCA | Esophageal carcinoma |
| HNSC | Head and neck squamous cell carcinoma |
| KICH | Kidney chromophobe |
| LUSC | Lung squamous cell carcinoma |
| PRAD | Prostate adenocarcinoma |
| STAD | Stomach adenocarcinoma |
| THCA | Thyroid carcinoma |
| UCEC | Uterine corpus endometrial carcinoma |
References
- Peter, M.E. Targeting of mrnas by multiple mirnas: The next step. Oncogene 2010, 29, 2161–2164. [Google Scholar] [CrossRef] [PubMed]
- Nunez, Y.O.; Truitt, J.M.; Gorini, G.; Ponomareva, O.N.; Blednov, Y.A.; Harris, R.A.; Mayfield, R.D. Positively correlated mirna-mrna regulatory networks in mouse frontal cortex during early stages of alcohol dependence. BMC Genom. 2013, 14, 725. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Z.; Hu, X.; Chen, Y. Biclustering of microarray data with mospo based on crowding distance. BMC Bioinform. 2009, 10, S9. [Google Scholar] [CrossRef] [PubMed]
- Tanay, A.; Sharan, R.; Shamir, R. Discovering statistically significant biclusters in gene expression data. Bioinformatics 2002, 18, S136–S144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Ding, L.; Baker, S.; Bai, J.M.; Rath, E.; Jiang, F.; Wu, J.; Jiang, H.; Stuart, G. Dissecting the biological relationship between TCGA miRNA and mRNA sequencing data using MMiRNA-Viewer. BMC Bioinform. 2016, 17, 336. [Google Scholar] [CrossRef] [PubMed]
- Oulas, A.; Karathanasis, N.; Louloupi, A.; Iliopoulos, I.; Kalantidis, K.; Poirazi, P. A new microrna target prediction tool identifies a novel interaction of a putative miRNA with CCND2. RNA Biol. 2012, 9, 1196–1207. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.; Bartel, D.P. Predicting effective microrna target sites in mammalian mrnas. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human microrna targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef]
- Vincent, D.B.; Jean-Loup, G.; Renaud, L.; Etienne, L. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [Green Version]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the american association for the study of liver diseases, american college of gastroenterology, and the american gastroenterological association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Kosir, R.; Rozman, D. Genomic aspects of nafld pathogenesis. Genomics 2013, 102, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-L.; Ming, Y.-N.; Zhang, J.-Y.; Chen, X.-Y.; Zeng, M.-D.; Mao, Y.-M. Gene-metabolite network analysis in different nonalcoholic fatty liver disease phenotypes. Exp. Mol. Med. 2017, 49, e283. [Google Scholar] [CrossRef] [PubMed]
- Ryaboshapkina, M.; Hammar, M. Human hepatic gene expression signature of non-alcoholic fatty liver disease progression, a meta-analysis. Sci. Rep. 2017, 7, 12361. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Tian, G.Y.; Song, Y.; Liu, Y.L.; Chen, Y.D.; Shi, J.P.; Yang, J. Characterization of transcriptional modules related to fibrosing-nafld progression. Sci. Rep. 2017, 7, 4748. [Google Scholar] [CrossRef] [PubMed]
- Soronen, J.; Yki-Jarvinen, H.; Zhou, Y.; Sadevirta, S.; Sarin, A.P.; Leivonen, M.; Sevastianova, K.; Perttila, J.; Laurila, P.P.; Sigruener, A.; et al. Novel hepatic micrornas upregulated in human nonalcoholic fatty liver disease. Physiol. Rep. 2016, 4, e12661. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Zhang, T.; Lou, G.; Xu, W.; Dong, F.; Chen, G.; Liu, Y. Plasma mir-17, mir-20a, mir-20b and mir-122 as potential biomarkers for diagnosis of nafld in type 2 diabetes mellitus patients. Life Sci. 2018, 208, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.-J.; Zhang, L.-P.; Duan, C.-Y.; Wang, B.; He, N.-N.; Abulimiti, P.; Lin, Y. The inhibition role of mir-22 in hepatocellular carcinoma cell migration and invasion via targeting cd147. Cancer Cell Int. 2017, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Y.; Ji, Z.; Wang, Z. Identification and differential expression of micrornas associated with fat deposition in the liver of wistar rats with nonalcoholic fatty liver disease. Gene 2016, 585, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.; Han, Y.; He, Q. Clusterprofiler: An r package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Malcolm, K.C.; Ross, A.H.; Qiu, R.G.; Symons, M.; Exton, J.H. Activation of rat liver phospholipase d by the small gtp-binding protein rhoa. J. Biol. Chem. 1994, 269, 25951–25954. [Google Scholar]
- Steller, E.J.; Raats, D.A.; Koster, J.; Rutten, B.; Govaert, K.M.; Emmink, B.L.; Snoeren, N.; van Hooff, S.R.; Holstege, F.C.; Maas, C.; et al. Pdgfrb promotes liver metastasis formation of mesenchymal-like colorectal tumor cells. Neoplasia 2013, 15, 204–217. [Google Scholar] [CrossRef]
- Wang, G.; Yang, X.; Li, C.; Cao, X.; Luo, X.; Hu, J. Pik3r3 induces epithelial-to-mesenchymal transition and promotes metastasis in colorectal cancer. Mol. Cancer Ther. 2014, 13, 1837–1847. [Google Scholar] [CrossRef]
- Hu, J.; Xia, X.; Cheng, A.; Wang, G.; Luo, X.; Reed, M.F.; Fojo, T.; Oetting, A.; Gong, J.; Yen, P.M. A peptide inhibitor derived from p55pik phosphatidylinositol 3-kinase regulatory subunit: A novel cancer therapy. Mol. Cancer Ther. 2008, 7, 3719. [Google Scholar] [CrossRef]
- Wu, X.; Chen, L.; Wang, X. Network biomarkers, interaction networks and dynamical network biomarkers in respiratory diseases. Clin. Transl. Med. 2014, 3, 16. [Google Scholar] [CrossRef]
- Lin, Z.H.; Xin, Y.N.; Dong, Q.J.; Wang, Q.; Jiang, X.J.; Zhan, S.H.; Sun, Y.; Xuan, S.Y. Performance of the aspartate aminotransferase-to-platelet ratio index for the staging of hepatitis c-related fibrosis: An updated meta-analysis. Hepatology 2011, 53, 726–736. [Google Scholar] [CrossRef]
- Sterling, R.K.; Lissen, E.; Clumeck, N.; Sola, R.; Correa, M.C.; Montaner, J.; Sulkowski, M.S.; Torriani, F.J.; Dieterich, D.T.; Thomas, D.L.; et al. Development of a simple noninvasive index to predict significant fibrosis in patients with hiv/hcv coinfection. Hepatology 2006, 43, 1317–1325. [Google Scholar] [CrossRef]
- Yang, B.; Li, M.; Tang, W.; Liu, W.; Zhang, S.; Chen, L.; Xia, J. Dynamic network biomarker indicates pulmonary metastasis at the tipping point of hepatocellular carcinoma. Nat. Commun. 2018, 9, 678. [Google Scholar] [CrossRef]
- Paul, S.; Lakatos, P.; Hartmann, A.; Schneider-Stock, R.; Vera, J. Identification of mirna-mrna modules in colorectal cancer using rough hypercuboid based supervised clustering. Sci. Rep. 2017, 7, 42809. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, W.; Xu, Y.; Li, C.; Wang, Y.; Yang, H.; Zhang, C.; Su, F.; Li, Y.; Li, X. Identification of subtype specific mirna-mrna functional regulatory modules in matched mirna-mrna expression data: Multiple myeloma as a case. Biomed. Res. Int. 2015, 2015, 15. [Google Scholar] [CrossRef]
- Seo, J.; Jin, D.; Choi, C.H.; Lee, H. Integration of microrna, mrna, and protein expression data for the identification of cancer-related micrornas. PLoS ONE 2017, 12, e0168412. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Types | Number of miRNA–mRNA Pairs with Inverse Correlations | Number of miRNA–mRNA Pairs with Inverse Correlations and Opposite Fold Change Between Tumor and Normal Samples |
|---|---|---|
| BLCA | 998 | 578 |
| BRCA | 20,661 | 10,101 |
| COAD | 82 | 55 |
| ESCA | 344 | 155 |
| HNSC | 3066 | 1601 |
| KICH | 1039 | 442 |
| KIRC | 10,749 | 6189 |
| KIRP | 6143 | 3190 |
| LIHC | 1426 | 659 |
| LUAD | 26,380 | 12,874 |
| LUSC | 265 | 171 |
| PRAD | 6972 | 3801 |
| STAD | 12,892 | 5108 |
| THCA | 1326 | 744 |
| UCEC | 408 | 214 |
| Total | 92,751 | 45,882 |
| Cancer Types | Total Number of Detected Clusters | Number of Detected Significant Clusters (FDR < 0.1) |
|---|---|---|
| BLCA | 28 | 2 |
| BRCA | 33 | 8 |
| COAD | 20 | 0 |
| ESCA | 42 | 0 |
| HNSC | 96 | 4 |
| KICH | 64 | 1 |
| KIRC | 51 | 8 |
| KIRP | 62 | 4 |
| LIHC | 114 | 1 |
| LUAD | 21 | 9 |
| LUSC | 39 | 2 |
| PRAD | 52 | 3 |
| STAD | 39 | 8 |
| THCA | 57 | 4 |
| UCEC | 70 | 0 |
| Total | 788 | 54 |
| Gene | miRNA |
|---|---|
| ITPKB | hsa-mir-106b |
| CD69 | hsa-mir-106b |
| EPHA4 | hsa-mir-106b |
| APOBEC3H | hsa-mir-106b |
| CYP2U1 | hsa-mir-106b |
| ZNFX1 | hsa-mir-106b |
| CNTNAP1 | hsa-mir-505 |
| EFCAB1 | hsa-mir-505 |
| BTG1 | hsa-mir-505 |
| HPRT1 | hsa-mir-505 |
| PAM | hsa-mir-505 |
| IRF2BP2 | hsa-mir-505 |
| FST | hsa-mir-505 |
| CLDN23 | hsa-mir-505 |
| SIN3A | hsa-mir-20b |
| XPR1 | hsa-mir-2355 |
| C7orf49 | hsa-mir-2355 |
| ZDHHC23 | hsa-mir-2355 |
| VANGL1 | hsa-mir-2355 |
| SSX2IP | hsa-mir-584 |
| DYNLT3 | hsa-mir-584 |
| ESR1 | hsa-mir-584 |
| ARL15 | hsa-mir-877 |
| MEST | hsa-mir-181d |
| TBCC | hsa-mir-374b |
| GUCY1A2 | hsa-mir-551b |
| SCO1 | hsa-mir-200b |
| CASC4 | hsa-mir-200b |
| FAM169A | hsa-mir-200b |
| UGGT1 | hsa-let-7b |
| PLEKHA6 | hsa-let-7b |
| ATP6V1C1 | hsa-let-7b |
| Gene | miRNA |
|---|---|
| DTNA | mir-122 |
| SMYD2 | mir-122 |
| IGF2 | mir-122 |
| KYNU | mir-122 |
| DBNDD1 | mir-122 |
| SYNCRIP | let-7c |
| KIF5B | let-7c |
| MGAT4A | let-7c |
| PDLIM2 | let-7c |
| LDHD | let-7c |
| PLCB1 | let-7c |
| BDH1 | let-7c |
| STXBP4 | let-7c |
| UGGT1 | let-7b |
| PLEKHA6 | let-7b |
| ATP6V1C1 | let-7b |
| CBX7 | mir-192 |
| ZC3H10 | mir-192 |
| RAB2A | mir-192 |
| TRIM66 | mir-192 |
| MYO1E | mir-192 |
| ING5 | mir-192 |
| SYAP1 | mir-192 |
| P2RX4 | mir-29a |
| ZNF286B | mir-29a |
| CNDP2 | mir-29a |
| GPR146 | mir-29a |
| BMF | mir-29a |
| SSTR2 | mir-29a |
| NLN | mir-29a |
| AMICA1 | mir-29a |
| SYNM | mir-29a |
| PRPF3 | mir-29a |
| CHST10 | mir-29a |
| ZNF160 | mir-29a |
| NDN | mir-29a |
| MTMR2 | mir-29a |
| ZNF431 | mir-29a |
| NAP1L1 | mir-29a |
| ATP6V0E2 | mir-29a |
| ATPAF1 | mir-29a |
| MORF4L1 | mir-29a |
| PRR3 | mir-29a |
| CPT2 | mir-29a |
| DNAJA3 | mir-29a |
| RIT1 | mir-29a |
| UCP3 | mir-29a |
| ZNF35 | mir-21 |
| WDR72 | mir-21 |
| KIAA1804 | mir-21 |
| LAMP2 | mir-21 |
| PFN2 | mir-21 |
| NFASC | mir-21 |
| FABP4 | mir-21 |
| C7 | mir-21 |
| STK3 | mir-21 |
| RASGRF1 | mir-132 |
| STK3 | mir-132 |
| PFN2 | mir-132 |
| MEST | mir-132 |
| NCALD | mir-132 |
| C9orf156 | mir-132 |
| LAMP2 | mir-99a |
| RCBTB1 | mir-99a |
| KPTN | mir-99a |
| RPS20 | mir-99a |
| ZDHHC18 | mir-99a |
| ABCB4 | mir-200c |
| PGAM1 | mir-200c |
| SCO1 | mir-200c |
| IGFBP2 | mir-145 |
| PRPF38A | mir-145 |
| CDK5RAP3 | mir-145 |
| RBMX | mir-145 |
| MGLL | mir-145 |
| Gene | miRNA |
|---|---|
| DGKQ | mir-140 |
| LPAR2 | mir-140 |
| PDGFRB | mir-186 |
| PIK3R3 | mir-151 |
| PIK3R3 | mir-148b |
| PIK3R3 | mir-589 |
| PTGFR | mir-107 |
| RAPGEF3 | mir-454 |
| RAPGEF3 | mir-93 |
| RAPGEF3 | mir-25 |
| RAPGEF3 | mir-186 |
| RAPGEF3 | mir-942 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, X.; Ding, L.; Liu, H.; Xu, Z.; Jiang, H.; Handelman, S.K.; Bai, Y. Identifying Interaction Clusters for MiRNA and MRNA Pairs in TCGA Network. Genes 2019, 10, 702. https://doi.org/10.3390/genes10090702
Dai X, Ding L, Liu H, Xu Z, Jiang H, Handelman SK, Bai Y. Identifying Interaction Clusters for MiRNA and MRNA Pairs in TCGA Network. Genes. 2019; 10(9):702. https://doi.org/10.3390/genes10090702
Chicago/Turabian StyleDai, Xinqing, Lizhong Ding, Hannah Liu, Zesheng Xu, Hui Jiang, Samuel K Handelman, and Yongsheng Bai. 2019. "Identifying Interaction Clusters for MiRNA and MRNA Pairs in TCGA Network" Genes 10, no. 9: 702. https://doi.org/10.3390/genes10090702