Anti-Colorectal Cancer Effects of Probiotic-Derived p8 Protein

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture
2.2. Construction of Codon-Optimized His-Tagged r-p8 Protein, and Expression and Purification in E. coli
2.3. Expression of Codon-Optimized r-p8 Protein in DLD-1 Cells
2.4. Cell Culture
2.5. Cell Proliferation Assay
2.6. Wound Healing Assay
2.7. ELISA Analysis
2.8. Western Blot Analysis
2.9. Immunocytochemistry Using ImageXpress® Micro Confocal Microscopy
2.10. Flow Cytometry Analysis
3. Result
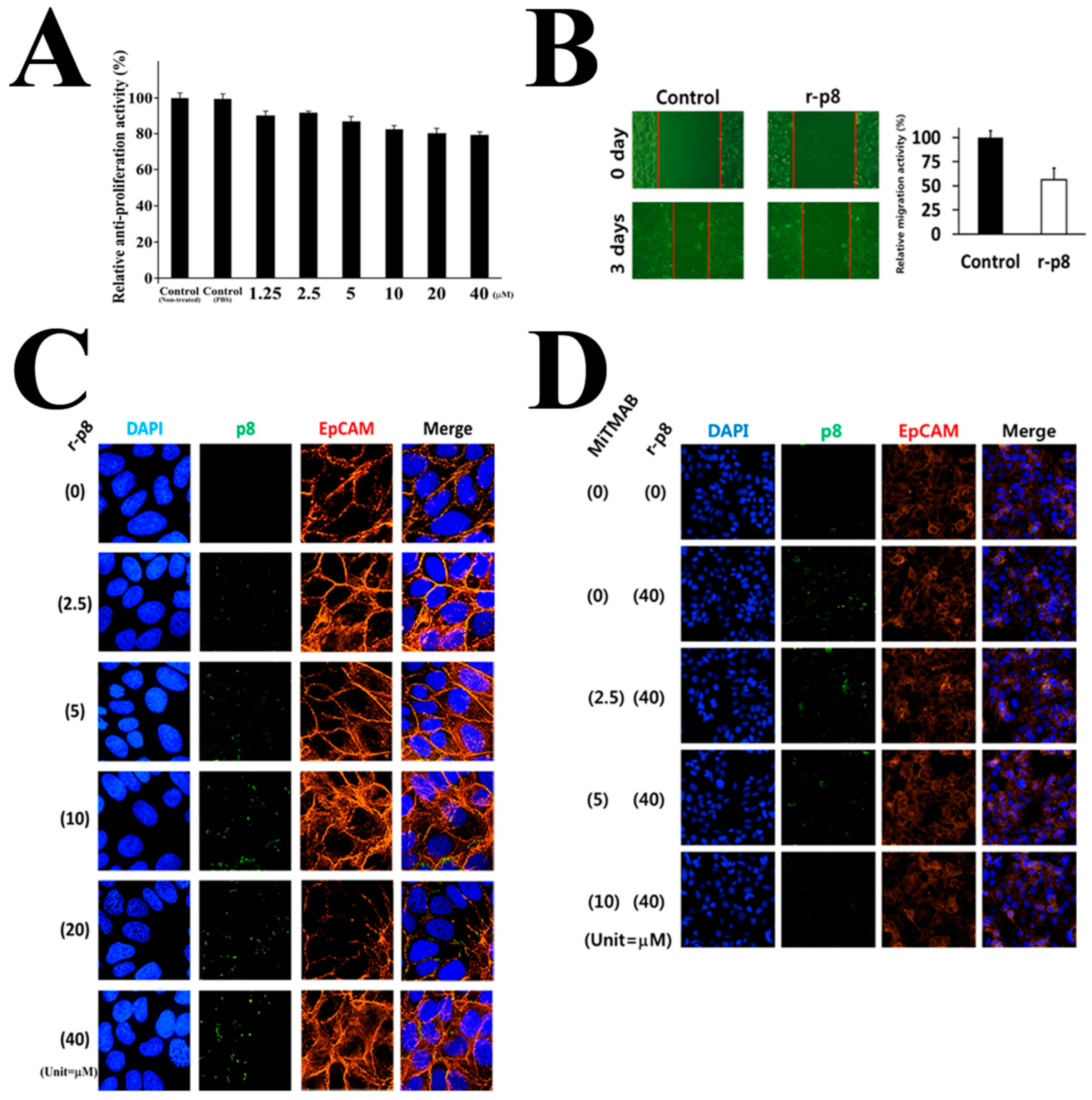
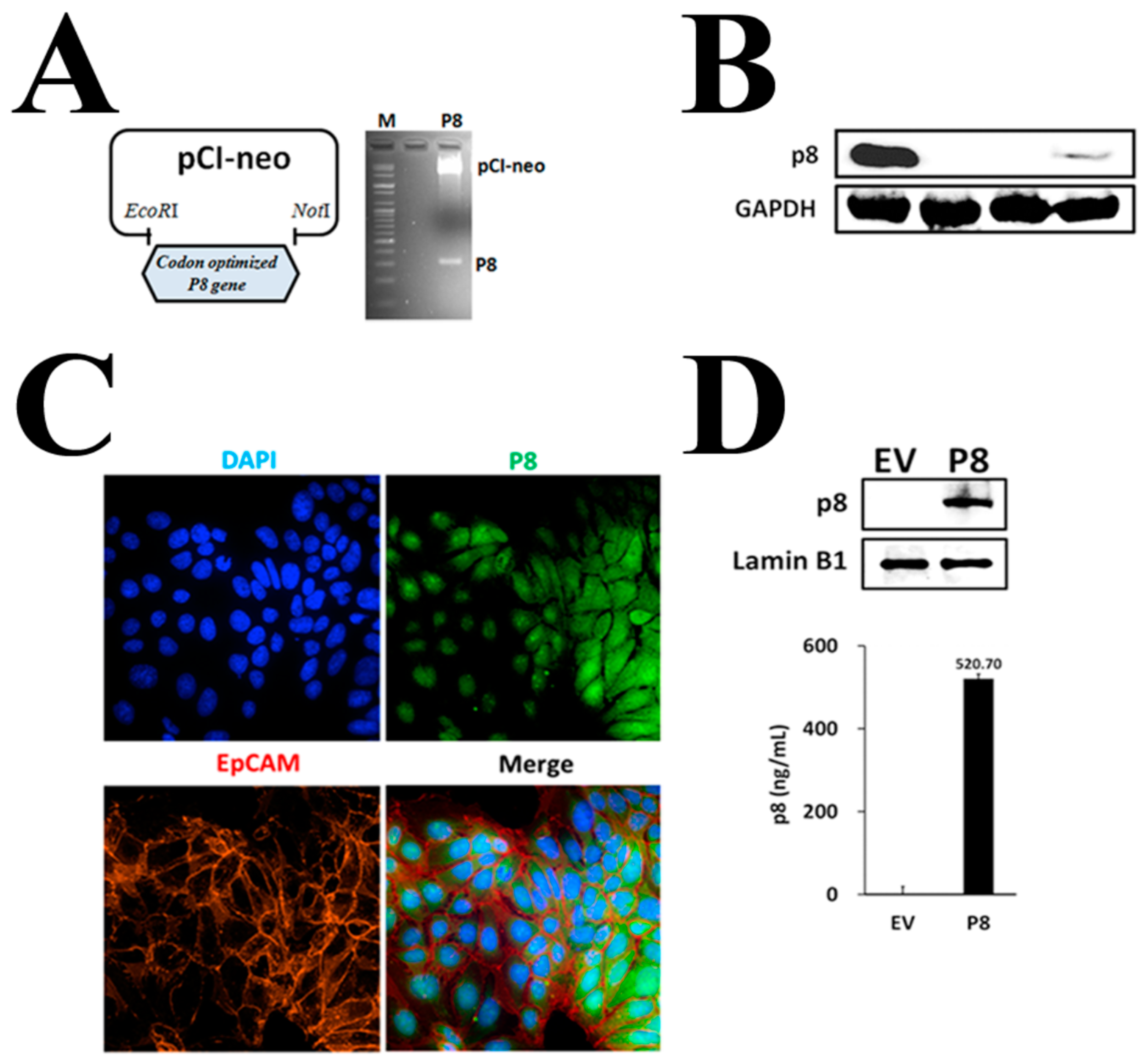
3.1. P8 Requires a Specific Delivery System
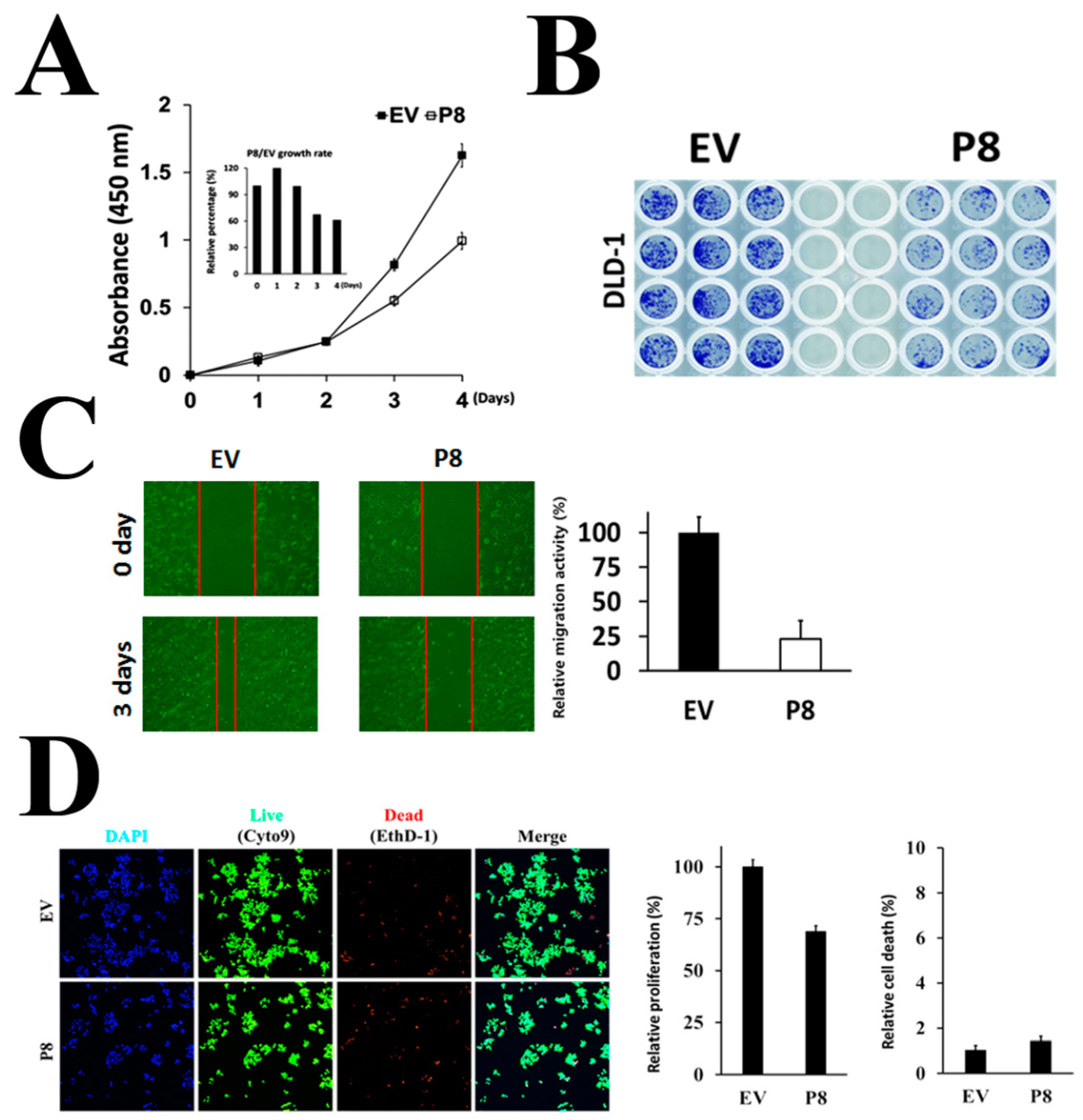
3.2. Endogenous p8 Expression Showed Markedly Enhanced Anti-Cancer Activity
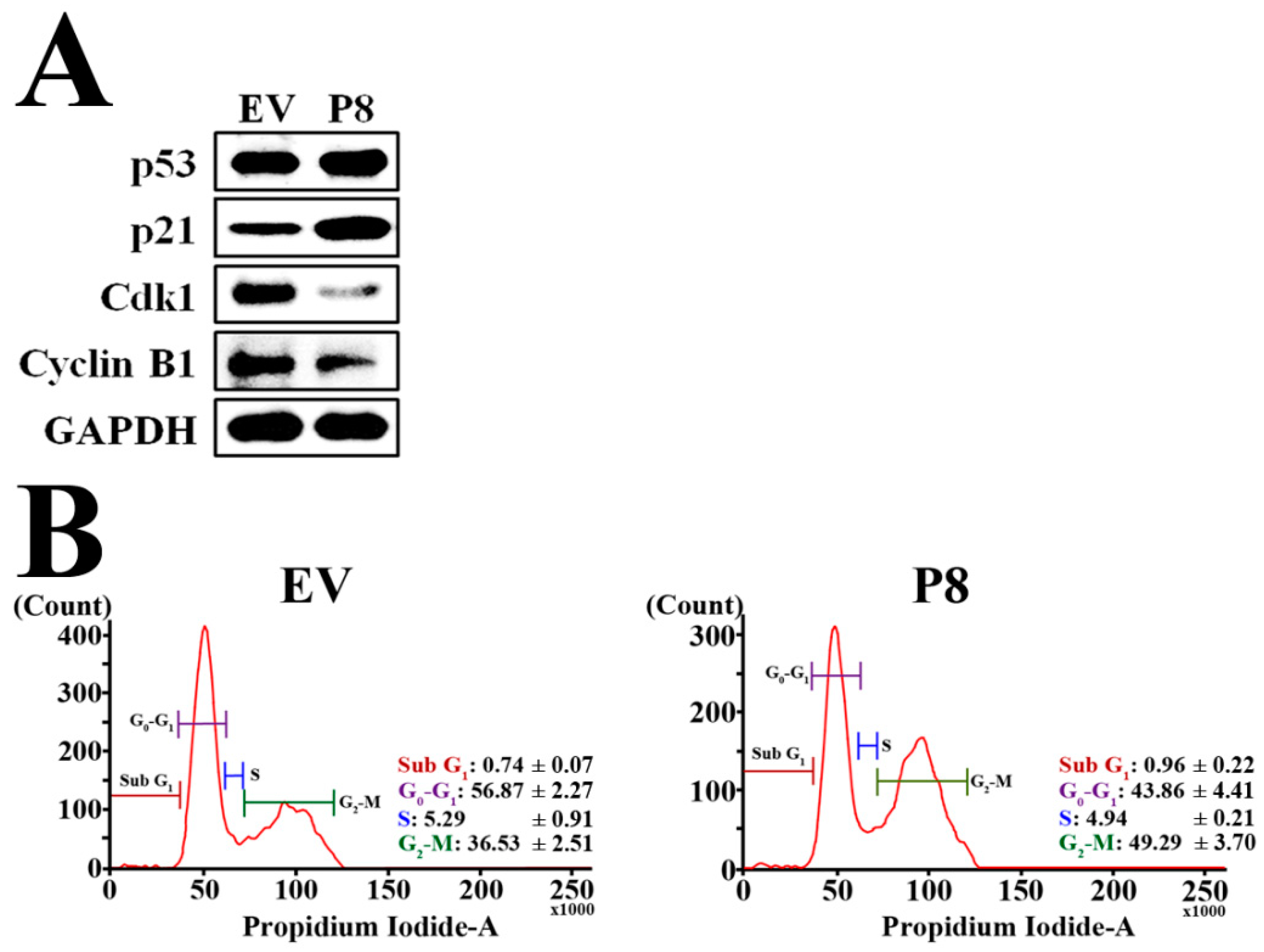
3.3. Effects of r-p8 on Anti-Cancer Signaling Pathways in DLD-1 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- SEER Stat Fact Sheets: Colon and Rectum Cancer. NCI. Available online: https://www.cancer.gov/publications/fact-sheets (accessed on 4 June 2019).
- Defining Cancer. National Cancer Institute, 17 September 2007. Available online: https://seer.cancer.gov/statistics/types.html (accessed on 4 June 2019).
- Colon Cancer Treatment (PDQ®). NCI. Available online: https://www.cancer.gov/types/colorectal/hp/colon-treatment-pdq (accessed on 4 June 2019).
- Zhang, L.; Ren, X.; Alt, E.; Bai, X.; Huang, S.; Xu, Z.; Lynch, P.M.; Moyer, M.P.; Wen, X.-F.; Wu, X. Chemoprevention of colorectal cancer by targeting APC-deficient cells for apoptosis. Nature 2010, 464, 1058–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, F.A. Development of Probiotics as Biologic Drugs. Clin. Infect. Dis. 2008, 46 (Suppl. 2), 125–127. [Google Scholar] [CrossRef]
- Mercenier, A.; Pavan, S.; Pot, B. Probiotics as Biotherapeutic Agents: Present Knowledge and Future Prospects. Curr. Pharm. Des. 2003, 9, 175–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seal, B.S.; Drider, D.; Oakley, B.B.; Brüssow, H.; Bikard, D.; Rich, J.O.; Miller, S.; Devillard, E.; Kwan, J.; Bertin, G.; et al. Microbial-derived products as potential new antimicrobials. Vet. Res. 2018, 49, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quigley, E.M. Gut microbiota and the role of probiotics in therapy. Curr. Opin. Pharmacol. 2011, 11, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Zhang, X.; Covasa, M. Emerging roles of lactic acid bacteria in protection against colorectal cancer. World J. Gastroenterol. 2014, 20, 7878–7886. [Google Scholar] [CrossRef]
- Sadeghi-Aliabadi, H.; Mohammadi, F.; Fazeli, H.; Mirlohi, M. Effects of Lactobacillus plantarum A7 with probiotic potential on colon cancer and normal cells proliferation in comparison with a commercial strain. Iran J. Basic Med. Sci. 2014, 17, 815–819. [Google Scholar]
- Widakowich, C.; de Castro, G., Jr.; de Azambuja, E.; Dinh, P.; Awada, A. Review: Side effects of approved molecular targeted therapies in solid cancers. Oncologist 2007, 12, 1443–1455. [Google Scholar] [CrossRef]
- Steidler, L.; Vandenbroucke, K. Genetically modified Lactococcus lactis: Novel tools for drug delivery. Int. J. Dairy Technol. 2006, 59, 140–146. [Google Scholar] [CrossRef]
- An, B.C.; Ryu, Y.; Yoon, Y.-S.; Choi, O.; Park, H.J.; Kim, T.Y.; Chung, M.J. Colorectal cancer therapy using a Pediococcus pentosaceus SL4 drug delivery system secreting lactic acid bacteria-derived protein p8. Moll. Cells. under review.
- Wang, S.; Noberini, R.; Stebbins, J.L.; Das, S.; Zhang, Z.; Wu, B.; Mitra, S.; Billet, S.; Fernandez, A.; Bhowmick, N.A.; et al. Targeted delivery of paclitaxel to EphA2-expressing cancer cells. Clin. Cancer Res. 2013, 19, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Sun, M. Martin Cline loses appeal on NIH grant. Science 1982, 218, 37. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Aebersold, P.; Cornetta, K.; Kasid, A.; Morgan, R.A.; Moen, R.; Karson, E.M.; Lotze, M.T.; Yang, J.C.; Topalian, S.L.; et al. Gene Transfer into Humans—Immunotherapy of Patients with Advanced Melanoma, Using Tumor-Infiltrating Lymphocytes Modified by Retroviral Gene Transduction. N. Engl. J. Med. 1990, 323, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Gene Therapy Clinical Trials Worldwide Database. J. Gene. Med. Wiley. Available online: http://www.abedia.com/wiley/phases.php (accessed on 4 June 2019).
- Pearson, S.; Jia, H.; Kandachi, K. China approves first gene therapy. Nat. Biotechnol. 2004, 22, 3–4. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine, Genomics Home Reference. What Is Gene Therapy? U.S. National Library of Medicine. Available online: https://ghr.nlm.nih.gov/primer/therapy/genetherapy (accessed on 4 June 2019).
- Vogelstein, B.; Hamilton, S.R.; Preisinger, A.C.; Leppert, M.; Smits, A.M.; Bos, J.L.; Fearon, E.R.; Kern, S.E. Genetic Alterations during Colorectal-Tumor Development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.J.; Seymour, L.W.; Maruta, F. Gene therapy for colorectal cancer. Expert. Opin. Biol. Ther. 2003, 3, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Lo, H.W.; Day, C.P.; Hung, M.C. Cancer-specific gene therapy. Adv. Genet. 2005, 54, 235–255. [Google Scholar]
- Dorer, D.E.; Nettelbeck, D.M. Targeting cancer by transcriptional control in cancer gene therapy and viral oncolysis. Adv. Drug Deliv. Rev. 2009, 61, 554–571. [Google Scholar] [CrossRef]
- Zhang, K.-X.; Jia, W.; Rennie, P.S. Bioengineered viral vectors for targeting and killing prostate cancer cells. Bioeng. Bugs 2010, 1, 92–96. [Google Scholar] [CrossRef] [Green Version]
- LaRocca, C.; Schlom, J. Viral Vector–based Therapeutic Cancer Vaccines. Cancer J. 2011, 17, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Ginn, S.L.; Edelstein, M.L.; Abedi, M.R.; Wixon, J.; Alexander, I.E.; Wixon, J. Gene therapy clinical trials worldwide to 2012—An update. J. Gene Med. 2013, 15, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, R.; Melguizo, C.; Prados, J.; Alvarez, P.J.; Caba, O.; Rodriguez-Serrano, F.; Hita-Contreras, F.; Aranega, A. New Gene Therapy Strategies for Cancer Treatment: A Review of Recent Patents. Recent Pat. Anti-Cancer Drug Discov. 2012, 7, 297–312. [Google Scholar] [CrossRef]
- Zhou, D.; Zhou, X.; Bian, A.; Li, H.; Chen, H.; Small, J.C.; Li, Y.; Giles-Davis, W.; Xiang, Z.; Ertl, H.C.J. An efficient method of directly cloning chimpanzee adenovirus as a vaccine vector. Nat. Protoc. 2010, 5, 1775–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.-L.; Zhou, D. Viral delivery for gene therapy against cell movement in cancer. Adv. Drug Deliv. Rev. 2011, 63, 671–677. [Google Scholar] [CrossRef]
- Meisenberg, C.; Gilbert, C.D.; Chalmers, A.; Haley, V.; Gollins, S.; Ward, E.S.; El-Khamisy, F.S. Clinical and cellular roles for TDP1 and TOP1 in modulating colorectal cancer response to irinotecan. Mol. Cancer Ther. 2015, 14, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, D.; Atkin, W.; Lenz, H.J.; Lynch, H.T.; Minsky, B.; Nordling, B.; Starling, N. Colorectal cancer. Lancet 2010, 375, 1030–1047. [Google Scholar] [CrossRef]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, J.S.; Astrow, A.B. Adjuvant therapy of colon cancer. Semin. Oncol. 2001, 28, 30–40. [Google Scholar] [CrossRef]
- Zhong, X.; Neumann, P.; Corbo, M.; Loh, E. Recent Advances in Biotherapeutics Drug Discovery and Development. In Drug Discovery and Development—Present and Future; IntechOpen: London, UK, 2011; p. 15. [Google Scholar]
- Hall, M.P. Biotransformation and In Vivo Stability of Protein Biotherapeutics: Impact on Candidate Selection and Pharmacokinetic Profiling. Drug Metab. Dispos. 2014, 42, 1873–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermiston, T.W.; Kirn, D. Genetically based therapeutics for cancer: Similarities and contrasts with traditional drug discovery and development. Mol. Ther. 2005, 11, 496–507. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Organism/Mapping | Codon-Optimized Sequences | Size (bp) |
|---|---|---|
| Original P8 | atggcaacagtagatcctgaaaagacattgtttctcgatgaaccaatgaacaaggtatttgactggagcaacagcgaagcacctgtacgtgatgcgctgtgggattattacatggaaaagaacagccgtgataccatcaagactgaagaagaaatgaaaccagtcctagacatgtccgacgatgaggtcaaagccctagcagaaaaggttctcaagaagtaa | 222 |
| E. coli cells/6×His-TEV-P8 (NdeI/EcoRI) | catatgagaggatcgcatcaccatcaccatcac-attacgatatcccaacgaccgaaaacctgtattttcagggatcc-atggcaacagtagatcctgaaaagacattgtttctcgatgaaccaatgaacaaggtatttgactggagcaacagcgaagcacctgtccgtgatgcgctgtgggattattacatggaaaagaacagccgtgatactatcaagactgaagaagaaatgaaaccagtcctagacatgtccgacgacgaggtcaaagccctagcagaaaaggttctcaagaagtaggaattc | 305 |
| DLD-1 cells/P8 (EcoRI/NotI) | gaattcatggctactgtcgacccagaaaaaaccctgttcttggacgaaccaatgaataaagtctttgattggtccaactctgaggccccggtacgggatgcgttgtgggattactacatggaaaaaaattccagggataccattaaaacagaagaagaaatgaagccagttctggacatgagtgacgacgaagtgaaagccctcgcggaaaaagttctcaagaaataggcggccgc | 236 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, B.C.; Hong, S.; Park, H.J.; Kim, B.-K.; Ahn, J.Y.; Ryu, Y.; An, J.H.; Chung, M.J. Anti-Colorectal Cancer Effects of Probiotic-Derived p8 Protein. Genes 2019, 10, 624. https://doi.org/10.3390/genes10080624
An BC, Hong S, Park HJ, Kim B-K, Ahn JY, Ryu Y, An JH, Chung MJ. Anti-Colorectal Cancer Effects of Probiotic-Derived p8 Protein. Genes. 2019; 10(8):624. https://doi.org/10.3390/genes10080624
Chicago/Turabian StyleAn, Byung Chull, Sunwoong Hong, Ho Jin Park, Bong-Kyu Kim, Jun Young Ahn, Yongku Ryu, Jae Hyung An, and Myung Jun Chung. 2019. "Anti-Colorectal Cancer Effects of Probiotic-Derived p8 Protein" Genes 10, no. 8: 624. https://doi.org/10.3390/genes10080624