Food Tracking Perspective: DNA Metabarcoding to Identify Plant Composition in Complex and Processed Food Products

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Commercial Processed Foods and Mock Herbal Mixture for Qualitative Analysis
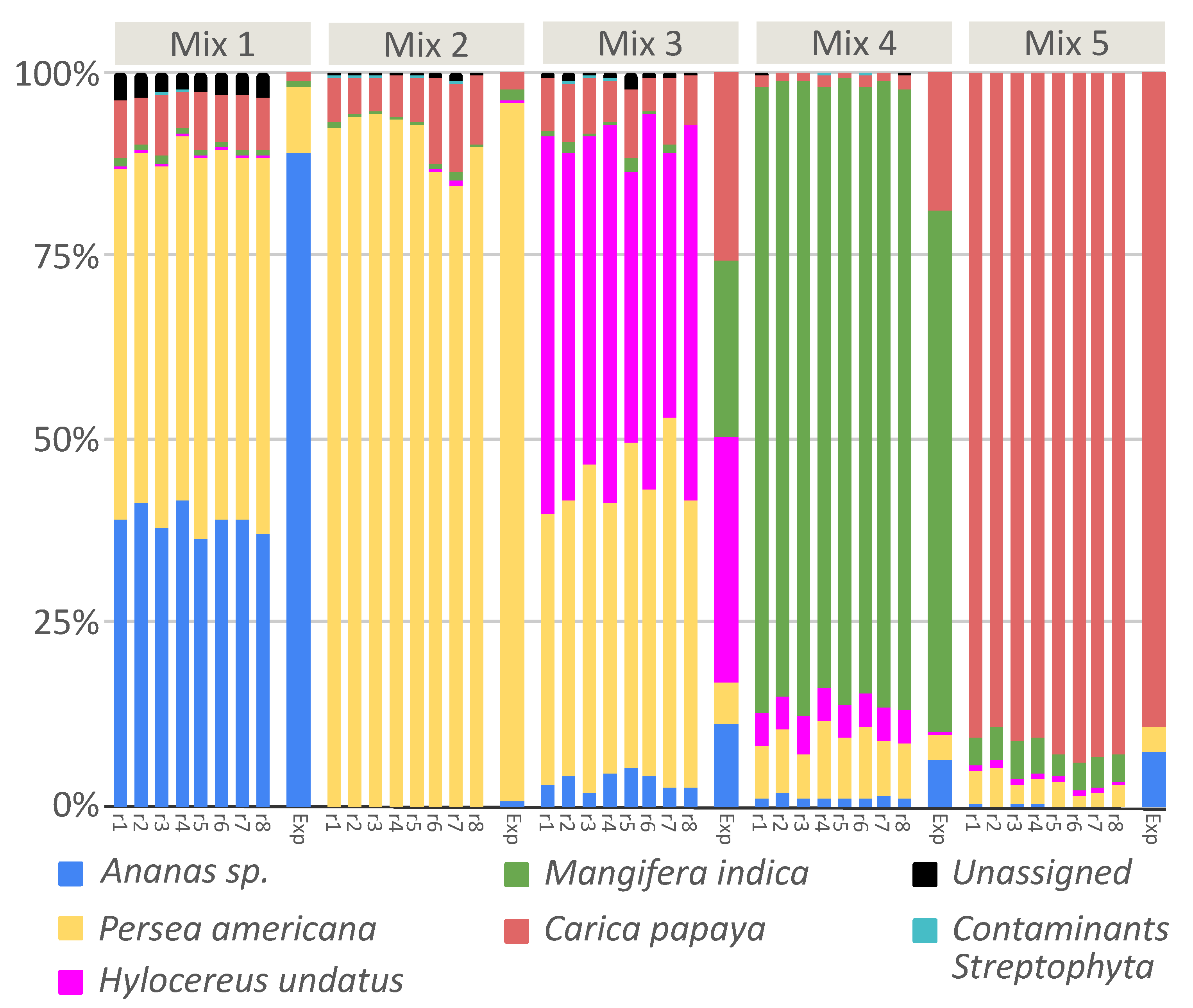
2.2. Fruit Mixtures Known in Composition and Quantity for Quantitative Analysis
2.3. DNA Extraction and qPCR
2.4. Sanger Sequencing
2.5. Libraries Preparation and Sequencing
2.6. Bioinformatics Analysis
3. Results and Discussion
3.1. Sequence Analysis
3.2. DNA Metabarcoding for Food Traceability
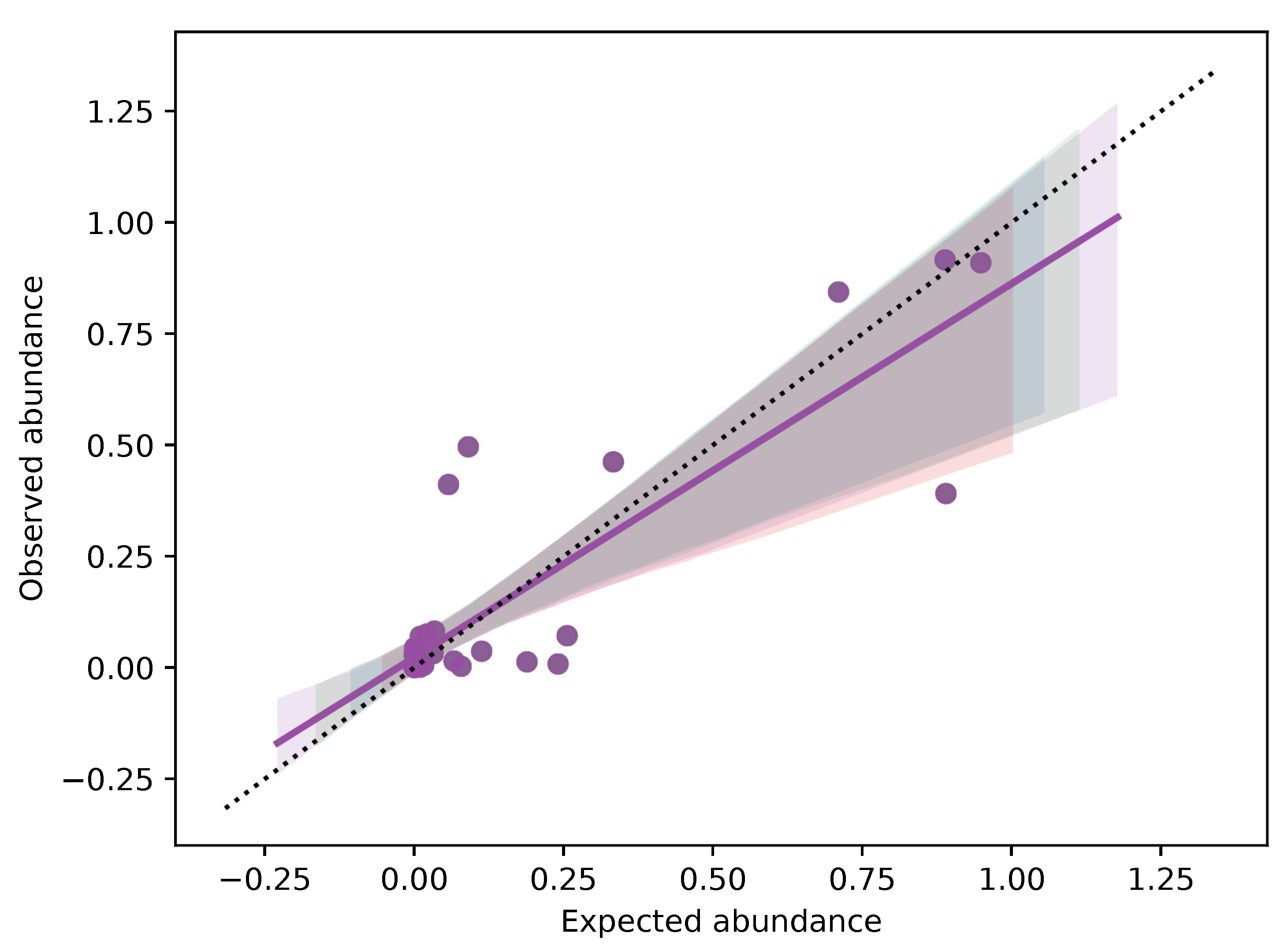
3.3. DNA Metabarcoding to Quantitatively Evaluate Food Composition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Valentini, P.; Galimberti, A.; Mezzasalma, V.; De Mattia, F.; Casiraghi, M.; Labra, M.; Pompa, P.P. DNA Barcoding Meets Nanotechnology: Development of a Universal Colorimetric Test for Food Authentication. Angew. Chem. Int. Ed. 2017, 56, 8094–8098. [Google Scholar] [CrossRef]
- Spencer, P.; Berman, F. Plant toxins and human health. In Food Safety: Contaminants and Toxins; Oxford University Press: Oxford, UK; CABI Publishing: Wallingford, Oxfordshire, UK, 2003; pp. 1–23. [Google Scholar]
- Barling, D.; Sharpe, R.; Lang, T. Traceability and ethical concerns in the UK wheat—Bread chain: From food safety to provenance to transparency. Int. J. Agric. Sustain. 2009, 7, 261–278. [Google Scholar] [CrossRef]
- Charlebois, S.; Sterling, B.; Haratifar, S.; Naing, S.K. Comparison of global food traceability regulations and requirements. Compr. Rev. Food Sci. Food Saf. 2014, 13, 1104–1123. [Google Scholar] [CrossRef]
- Lo, Y.T.; Shaw, P.C. DNA-based techniques for authentication of processed food and food supplements. Food Chem. 2018, 240, 767–774. [Google Scholar] [CrossRef]
- De Mattia, F.; Bruni, I.; Galimberti, A.; Cattaneo, F.; Casiraghi, M.; Labra, M. A comparative study of different DNA barcoding markers for the identification of some members of Lamiacaea. Food Res. Int. 2011, 44, 693–702. [Google Scholar] [CrossRef]
- Galimberti, A.; Sandionigi, A.; Bruno, A.; Bruni, I.; Barbuto, M.; Casiraghi, M.; Labra, M. Towards a universal molecular approach for the quality control of new foodstuffs. In Advances in Food Biotechnology; Wiley Blackwell: New York, NY, USA, 2015; p. 37. [Google Scholar]
- Bruni, I.; Galimberti, A.; Caridi, L.; Scaccabarozzi, D.; De Mattia, F.; Casiraghi, M.; Labra, M. A DNA barcoding approach to identify plant species in multiflower honey. Food Chem. 2015, 170, 308–315. [Google Scholar] [CrossRef]
- Barbuto, M.; Galimberti, A.; Ferri, E.; Labra, M.; Malandra, R.; Galli, P.; Casiraghi, M. DNA barcoding reveals fraudulent substitutions in shark seafood products: The Italian case of “palombo” (Mustelus spp.). Food Res. Int. 2010, 43, 376–381. [Google Scholar] [CrossRef]
- de Boer, H.J.; Ichim, M.C.; Newmaster, S.G. DNA barcoding and pharmacovigilance of herbal medicines. Drug Saf. 2015, 38, 611–620. [Google Scholar] [CrossRef]
- Galimberti, A.; Bruno, A.; Mezzasalma, V.; De Mattia, F.; Bruni, I.; Labra, M. Emerging DNA-based technologies to characterize food ecosystems. Food Res. Int. 2015, 69, 424–433. [Google Scholar] [CrossRef]
- Staats, M.; Arulandhu, A.J.; Gravendeel, B.; Holst-Jensen, A.; Scholtens, I.; Peelen, T.; Prins, T.W.; Kok, E. Advances in DNA metabarcoding for food and wildlife forensic species identification. Anal. Bioanal. Chem. 2016, 408, 4615–4630. [Google Scholar] [CrossRef]
- Raclariu, A.C.; Heinrich, M.; Ichim, M.C.; de Boer, H. Benefits and limitations of DNA barcoding and metabarcoding in herbal product authentication. Phytochem. Anal. 2018, 29, 123–128. [Google Scholar] [CrossRef]
- Hrnčírová, Z.; Bergerová, E.; Siekel, P. Effects of technological treatment on DNA degradation in selected food matrices of plant origin. J. Food Nutr. Res. 2008, 47, 23–28. [Google Scholar]
- Gryson, N. Effect of food processing on plant DNA degradation and PCR-based GMO analysis: A review. Anal. Bioanal. Chem. 2010, 396, 2003–2022. [Google Scholar] [CrossRef]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef]
- Elbrecht, V.; Leese, F. Can DNA-based ecosystem assessments quantify species abundance? Testing primer bias and biomass—Sequence relationships with an innovative metabarcoding protocol. PLoS ONE 2015, 10, e0130324. [Google Scholar] [CrossRef]
- Deagle, B.E.; Jarman, S.N.; Coissac, E.; Pompanon, F.; Taberlet, P. DNA metabarcoding and the cytochrome c oxidase subunit I marker: Not a perfect match. Biol. Lett. 2014, 10, 20140562. [Google Scholar] [CrossRef]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR inhibitors–occurrence, properties and removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef]
- Ficetola, G.F.; Taberlet, P.; Coissac, E. How to limit false positives in environmental DNA and metabarcoding? Mol. Ecol. Resour. 2016, 16, 604–607. [Google Scholar] [CrossRef]
- Taberlet, P.; Coissac, E.; Pompanon, F.; Gielly, L.; Miquel, C.; Valentini, A.; Vermat, T.; Corthier, G.; Brochmann, C.; Willerslev, E. Power and limitations of the chloroplast trn L (UAA) intron for plant DNA barcoding. Nucleic Acids Res. 2006, 35, e14. [Google Scholar] [CrossRef]
- Faria, M.; Magalhães, A.; Nunes, M.; Oliveira, M. High resolution melting of trnL amplicons in fruit juices authentication. Food Control 2013, 33, 136–141. [Google Scholar] [CrossRef]
- Yang, Y.; Xie, B.; Yan, J. Application of next-generation sequencing technology in forensic science. Genom. Proteom. Bioinform. 2014, 12, 190–197. [Google Scholar] [CrossRef]
- Young, J.M.; Weyrich, L.S.; Cooper, A. Forensic soil DNA analysis using high-throughput sequencing: A comparison of four molecular markers. Forensic Sci. Int. Genet. 2014, 13, 176–184. [Google Scholar] [CrossRef]
- Kartzinel, T.R.; Chen, P.A.; Coverdale, T.C.; Erickson, D.L.; Kress, W.J.; Kuzmina, M.L.; Rubenstein, D.I.; Wang, W.; Pringle, R.M. DNA metabarcoding illuminates dietary niche partitioning by African large herbivores. Proc. Natl. Acad. Sci. USA 2015, 112, 8019–8024. [Google Scholar] [CrossRef]
- Heidarbeigi, K.; Mohtasebi, S.S.; Foroughirad, A.; Ghasemi-Varnamkhasti, M.; Rafiee, S.; Rezaei, K. Detection of adulteration in saffron samples using electronic nose. Int. J. Food Prop. 2015, 18, 1391–1401. [Google Scholar] [CrossRef]
- Bruno, A.; Sandionigi, A.; Galimberti, A.; Siani, E.; Labra, M.; Cocuzza, C.; Ferri, E.; Casiraghi, M. One step forwards for the routine use of high-throughput DNA sequencing in environmental monitoring: An efficient and standardizable method to maximize the detection of environmental bacteria. MicrobiologyOpen 2017, 6, e00421. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. QIIME 2: Reproducible, Interactive, Scalable, and Extensible Microbiome Data Science. PeerJ Preprints 2018. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Edgar, R.C.; Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 2015, 31, 3476–3482. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Särkinen, T.; Staats, M.; Richardson, J.E.; Cowan, R.S.; Bakker, F.T. How to open the treasure chest? Optimising DNA extraction from herbarium specimens. PLoS ONE 2012, 7, e43808. [Google Scholar] [CrossRef]
- Sahu, S.K.; Thangaraj, M.; Kathiresan, K. DNA extraction protocol for plants with high levels of secondary metabolites and polysaccharides without using liquid nitrogen and phenol. ISRN Mol. Biol. 2012, 2012, 205049. [Google Scholar] [CrossRef]
- Burns, M.; Wiseman, G.; Knight, A.; Bramley, P.; Foster, L.; Rollinson, S.; Damant, A.; Primrose, S. Measurement issues associated with quantitative molecular biology analysis of complex food matrices for the detection of food fraud. Analyst 2016, 141, 45–61. [Google Scholar] [CrossRef]
- Balech, B.; Sandionigi, A.; Manzari, C.; Trucchi, E.; Tullo, A.; Licciulli, F.; Grillo, G.; Sbisà, E.; De Felici, S.; Saccone, C.; et al. Tackling critical parameters in metazoan meta-barcoding experiments: A preliminary study based on coxI DNA barcode. PeerJ 2018, 6, e4845. [Google Scholar] [CrossRef]
- Thudi, M.; Li, Y.; Jackson, S.A.; May, G.D.; Varshney, R.K. Current state-of-art of sequencing technologies for plant genomics research. Brief. Funct. Genom. 2012, 11, 3–11. [Google Scholar] [CrossRef]
- Piñol, J.; Senar, M.A.; Symondson, W.O. The choice of universal primers and the characteristics of the species mixture determine when DNA metabarcoding can be quantitative. Mol. Ecol. 2019, 28, 407–419. [Google Scholar] [CrossRef]
- Bista, I.; Carvalho, G.R.; Tang, M.; Walsh, K.; Zhou, X.; Hajibabaei, M.; Shokralla, S.; Seymour, M.; Bradley, D.; Liu, S.; et al. Performance of amplicon and shotgun sequencing for accurate biomass estimation in invertebrate community samples. Mol. Ecol. Resour. 2018, 18, 1020–1034. [Google Scholar] [CrossRef]
- Elbrecht, V.; Vamos, E.E.; Steinke, D.; Leese, F. Estimating intraspecific genetic diversity from community DNA metabarcoding data. PeerJ 2018, 6, e4644. [Google Scholar] [CrossRef]
- Leray, M.; Yang, J.Y.; Meyer, C.P.; Mills, S.C.; Agudelo, N.; Ranwez, V.; Boehm, J.T.; Machida, R.J. A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: Application for characterizing coral reef fish gut contents. Front. Zool. 2013, 10, 34. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| trnL g | GGGCAATCCTGAGCCAA |
| trnL h | CCATTGAGTCTCTGCACCTATC |
| trnL c | CGAAATCGGTAGACGCTACG |
| DNA Metabarcoding | Not Declared Specimen | ||
|---|---|---|---|
| Sample | Declared Species Composition | Composition (Vsearch) | Contaminants |
| (>0.3%) | (False Positive) | ||
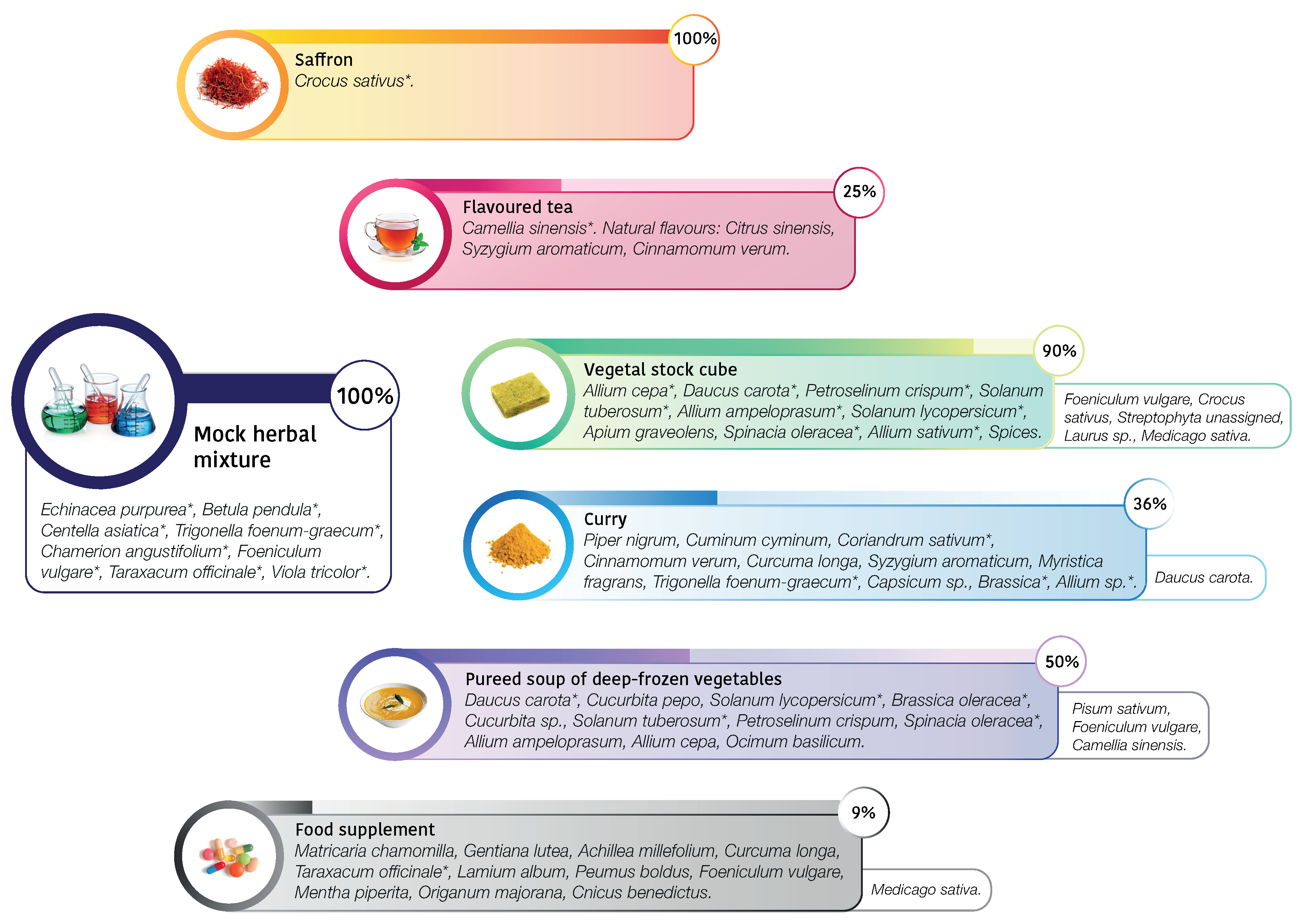
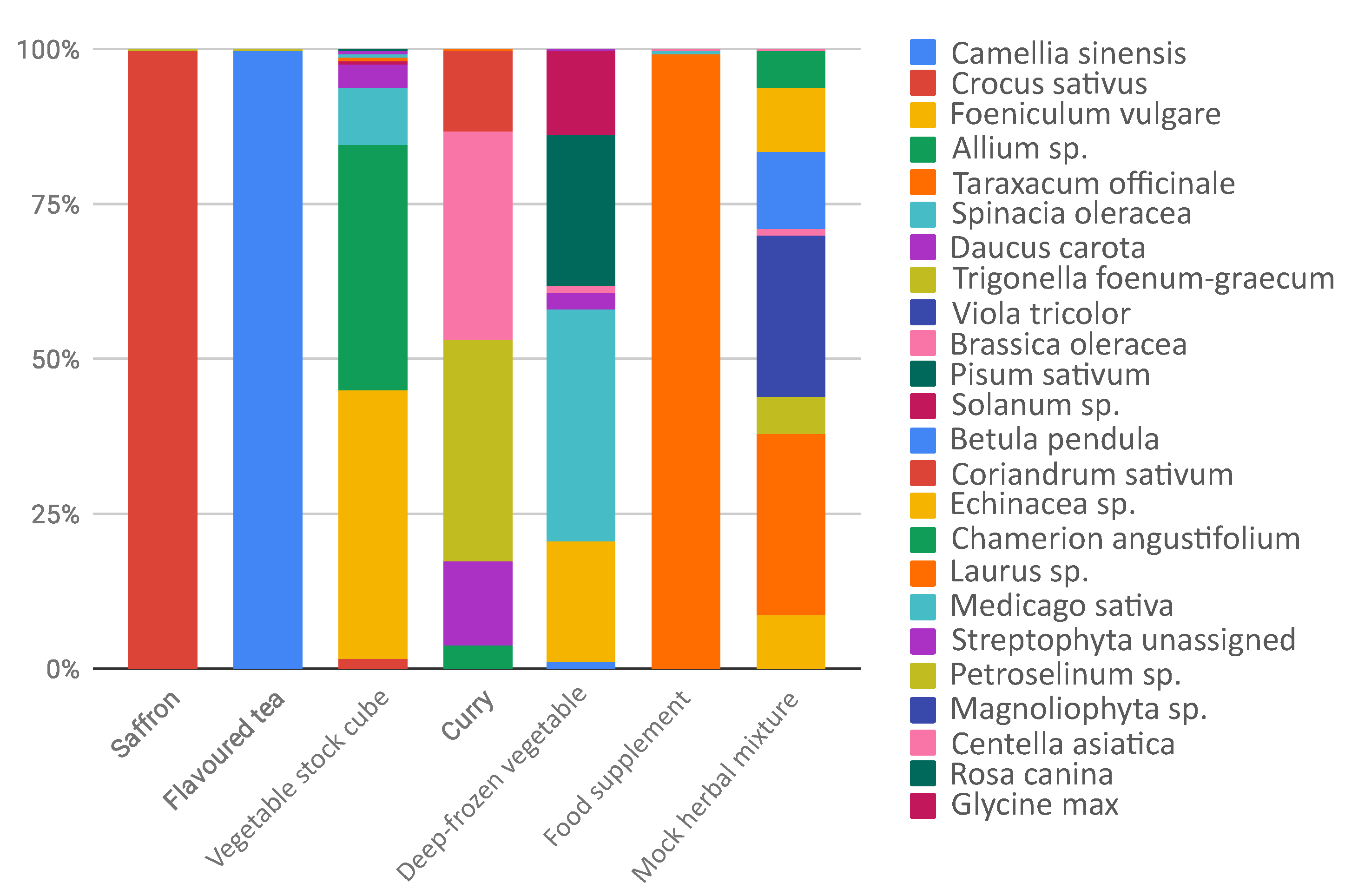
| saffron | Crocus sativus | Crocus sativus (99.8%) | |
| flavoured tea | Camellia sinensis | Camellia sinensis (99.7%) | |
| natural flavours: | |||
| Citrus sinensis | |||
| Syzygium aromaticum | |||
| Cinnamomum verum | |||
| vegetable stock cube | Allium cepa (1.9%) | Allium sp. (39.3%) | Foeniculum vulgare (43%) |
| Daucus carota (1%) | Spinacia oleracea (9.3%) | Crocus sativus (2.1%) | |
| Petroselinum crispum (0.5%) | Daucus carota (3.9%) | Streptophyta unassigned (0.5%) | |
| Solanum tuberosum (0.3%) | Petroselinum sp. (0.3%) | Laurus sp. (0.5%) | |
| Allium ampeloprasum (0.2%) | Solanum sp. (0.3%) | Medicago sativa (0.3%) | |
| Solanum lycopersicum (0.1%) | |||
| Apium graveolens (0.1%) | |||
| Spinacia oleracea (0.1%) | |||
| Allium sativum (0.1%) | |||
| spices | |||
| curry | Piper nigrum | Trigonella foenum-graecum (36%) | Daucus carota (13.4%) |
| Cuminum cyminum | Brassica sp. (33.3%) | ||
| Coriandrum sativum | Coriandrum sativum (13.2%) | ||
| Cinnamomum verum | Allium sp. (4%) | ||
| Curcuma longa | |||
| Syzygium aromaticum | |||
| Myristica fragrans | |||
| Trigonella foenum-graecum | |||
| Capsicum sp. | |||
| Brassica sp. | |||
| Allium sp. | |||
| deep-frozen vegetables | Daucus carota | Spinacia oleracea(37.2%) | Pisum sativum (24.8%) |
| Cucurbita pepo | Solanum sp. (13.3%) | Foeniculum vulgare (19.8%) | |
| Solanum lycopersicum | Daucus carota (2.6%) | Camellia sinensis (1.1%) | |
| Brassica oleracea | Brassica oleracea (0.9%) | ||
| Cucurbita sp. | |||
| Solanum tuberosum | |||
| Petroselinum crispum | |||
| Spinacia oleracea | |||
| Allium ampeloprasum | |||
| Allium cepa | |||
| Ocimum basilicum | |||
| food supplement | Matricaria chamomilla | Taraxacum officinale (99.2%) | Medicago sativa (0.4%) |
| Gentiana lutea | |||
| Achillea millefolium | |||
| Curcuma longa | |||
| Taraxacum officinale | |||
| Lamium album | |||
| Peumus boldus | |||
| Foeniculum vulgare | |||
| Mentha piperita | |||
| Origanum majorana | |||
| Cnicus benedictus | |||
| mock herbal mixture | Echinacea purpurea | Taraxacum officinale (29.3%) | |
| Betula pendula | Viola tricolor (26.3%) | ||
| Centella asiatica | Betula pendula (12.8%) | ||
| Trigonella foenum-graecum | Echinacea sp. (9.9%) | ||
| Chamerion angustifolium | Foeniculum vulgare (8.6%) | ||
| Foeniculum vulgare | Chamerion angustifolium (6%) | ||
| Taraxacum officinale | Trigonella foenum-graecum(5.8%) | ||
| Viola tricolor | Centella asiatica (0.4%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruno, A.; Sandionigi, A.; Agostinetto, G.; Bernabovi, L.; Frigerio, J.; Casiraghi, M.; Labra, M. Food Tracking Perspective: DNA Metabarcoding to Identify Plant Composition in Complex and Processed Food Products. Genes 2019, 10, 248. https://doi.org/10.3390/genes10030248
Bruno A, Sandionigi A, Agostinetto G, Bernabovi L, Frigerio J, Casiraghi M, Labra M. Food Tracking Perspective: DNA Metabarcoding to Identify Plant Composition in Complex and Processed Food Products. Genes. 2019; 10(3):248. https://doi.org/10.3390/genes10030248
Chicago/Turabian StyleBruno, Antonia, Anna Sandionigi, Giulia Agostinetto, Lorenzo Bernabovi, Jessica Frigerio, Maurizio Casiraghi, and Massimo Labra. 2019. "Food Tracking Perspective: DNA Metabarcoding to Identify Plant Composition in Complex and Processed Food Products" Genes 10, no. 3: 248. https://doi.org/10.3390/genes10030248