Challenges in the Integration of Omics and Non-Omics Data

 , , , and
, , , and
Abstract
:
1. Introduction
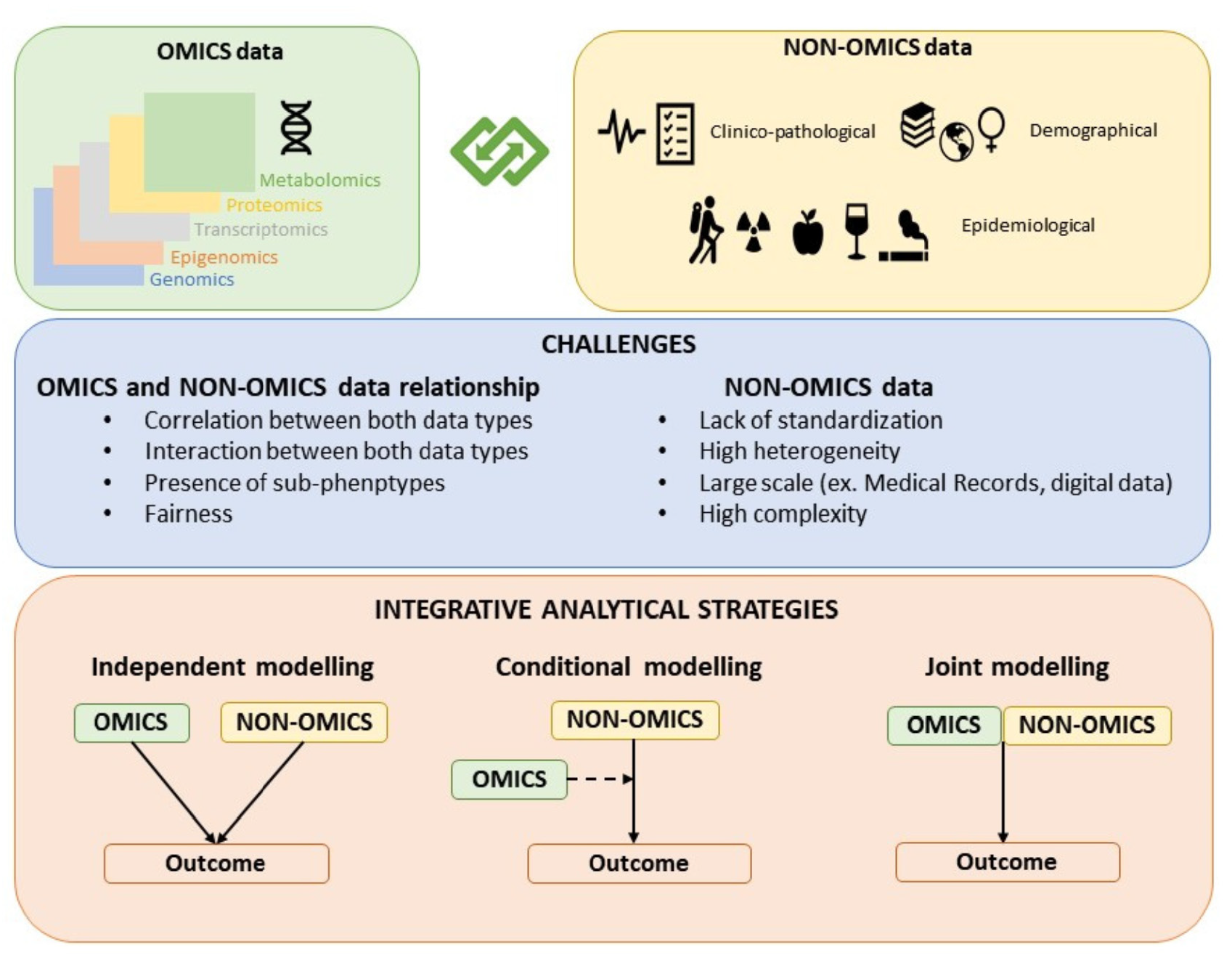
2. Challenges in Integrating Omics and Non-Omics Data
2.1. Challenges Due to the Nature of Non-Omics Data
2.1.1. Non-Omics Data Are Complex and Heterogeneously and Subjectively Defined
2.1.2. Heterogeneity Across Non-Omics Data
2.1.3. Large Scale Non-Omics Data
2.2. Challenges Due to the Relationship between Non-Omics and Omics Data
2.2.1. Ascertainment Bias
2.2.2. Interactions between Omics and Non-Omics Data
2.3. Other Challenges
2.3.1. Fairness
2.3.2. Presence of Subphenotypes
3. Integrative Analytical Strategies
3.1. Independent Modeling Approach
3.2. Conditional Modeling Approach
3.3. Joint Modeling Approach
4. Attempts of OnO Data Integration in Clinical and Epidemiological Studies
4.1. Study Objective
4.2. Study Outcome
4.3. Omics Data
4.4. Non-Omics Data
4.5. Integrative Analytical Strategies
4.6. OnO Data Integrative Models Performance
5. Recommended Integration Strategies
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Civelek, M.; Lusis, A.J. Systems genetics approaches to understand complex traits. Nat. Rev. Genet. 2014, 15, 34–48. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, V.N.; Lingjærde, O.C.; Russnes, H.G.; Vollan, H.K.M.; Frigessi, A.; Børresen-Dale, A.L. Principles and methods of integrative genomic analyses in cancer. Nat. Rev. Cancer 2014, 14, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Shen, R.; Olshen, A.B.; Ladanyi, M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics 2009, 25, 2906–2912. [Google Scholar] [CrossRef]
- Pineda, S.; Gomez-Rubio, P.; Picornell, A.; Bessonov, K.; Márquez, M.; Kogevinas, M.; Real, F.X.; Van Steen, K.; Malats, N. Framework for the Integration of Genomics, Epigenomics and Transcriptomics in Complex Diseases. Hum. Hered. 2015, 79, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Brandão, M.; Pondé, N.; Piccart-Gebhart, M. MammaprintTM: A comprehensive review. Futur. Oncol. 2019, 15, 207–224. [Google Scholar] [CrossRef]
- Wallden, B.; Ferree, S.; Ravi, H.; Dowidar, N.; Hood, T.; Danaher, P.; Mashadi-Hossein, A.; Wright, G.; Schaper, C.; Justin, J. Development of the molecular diagnostic (MDx) DLBCL Lymphoma Subtyping Test (LST) on the nCounter Analysis System. J. Clin. Oncol. 2015, 33. [Google Scholar]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Analyses of Cohorts of Twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef]
- Salathé, M.; Bengtsson, L.; Bodnar, T.J.; Brewer, D.D.; Brownstein, J.S.; Buckee, C.; Campbell, E.M.; Cattuto, C.; Khandelwal, S.; Mabry, P.L.; et al. Digital epidemiology. PLoS Comput. Biol. 2012, 8, 1–5. [Google Scholar] [CrossRef]
- Jette, A.M. Measuring subjective clinical outcomes. Phys. Ther. 1989, 69, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Tugwell, P.; Bombardier, C. A methodologic framework for developing and selecting endpoints in clinical trials. J. Rheumatol. 1982, 9, 758–762. [Google Scholar] [PubMed]
- Balliu, B.; Tsonaka, R.; Boehringer, S.; Houwing-Duistermaat, J. A Retrospective Likelihood Approach for Efficient Integration of Multiple Omics Factors in Case-Control Association Studies. Genet. Epidemiol. 2015, 39, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Spiegl-Kreinecker, S.; Lötsch, D.; Ghanim, B.; Pirker, C.; Mohr, T.; Laaber, M.; Weis, S.; Olschowski, A.; Webersinke, G.; Pichler, J.; et al. Prognostic quality of activating TERT promoter mutations in glioblastoma: Interaction with the rs2853669 polymorphism and patient age at diagnosis. Neuro. Oncol. 2015, 17, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Boks, M.P.; Derks, E.M.; Weisenberger, D.J.; Strengman, E.; Janson, E.; Sommer, I.E.; Kahn, R.S.; Ophoff, R.A. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS ONE 2009, 4, e6767. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.M.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef]
- Marchini, J.; Cardon, L.R.; Phillips, M.S.; Donnelly, P. The effects of human population structure on large genetic association studies. Nat. Genet. 2004, 36, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Van de Geer, J.P. Linear relations among k sets of variables. Psychometrika 1984, 49, 79–94. [Google Scholar] [CrossRef]
- López de Maturana, E.; Picornell, A.; Masson-Lecomte, A.; Kogevinas, M.; Márquez, M.; Carrato, A.; Tardón, A.; Lloreta, J.; García-Closas, M.; Silverman, D.; et al. Prediction of non-muscle invasive bladder cancer outcomes assessed by innovative multimarker prognostic models. BMC Cancer 2016, 16, 351. [Google Scholar] [CrossRef]
- Gevaert, O.; De Smet, F.; Timmerman, D.; Moreau, Y.; De Moor, B. Predicting the prognosis of breast cancer by integrating clinical and microarray data with Bayesian networks. Bioinformatics 2006, 22, e184–e190. [Google Scholar] [CrossRef] [PubMed]
- Nevins, J.R.; Huang, E.S.; Dressman, H.; Pittman, J.; Huang, A.T.; West, M. Towards integrated clinico-genomic models for personalized medicine: combining gene expression signatures and clinical factors in breast cancer outcomes prediction. Hum. Mol. Genet. 2003, 12, R153–R157. [Google Scholar] [CrossRef] [PubMed]
- Pittman, J.; Huang, E.; Dressman, H.; Horng, C.-F.; Cheng, S.H.; Tsou, M.-H.; Chen, C.-M.; Bild, A.; Iversen, E.S.; Huang, A.T.; et al. Integrated modeling of clinical and gene expression information for personalized prediction of disease outcomes. Proc. Natl. Acad. Sci. USA 2004, 101, 8431–8436. [Google Scholar] [CrossRef]
- Bøvelstad, H.M.; Nygård, S.; Borgan, Ø. Survival prediction from clinico-genomic models—A comparative study. BMC Bioinform. 2009, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Bazzoli, C.; Lambert-Lacroix, S. Classification based on extensions of LS-PLS using logistic regression: Application to clinical and multiple genomic data. BMC Bioinform. 2018, 19, 314. [Google Scholar] [CrossRef]
- Nygård, S.; Borgan, Ø.; Lingjærde, O.C.; Størvold, H.L. Partial least squares Cox regression for genome-wide data. Lifetime Data Anal. 2008, 14, 179–185. [Google Scholar] [CrossRef]
- Hoerl, A.E.; Kennard, R.W. Ridge Regression: Biased Estimation for Nonorthogonal Problems. Technometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Selection and Shrinkage via the Lasso. J. R. Stat. Soc. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Binder, H.; Schumacher, M. Allowing for mandatory covariates in boosting estimation of sparse high-dimensional survival models. BMC Bioinform. 2008, 9, 14. [Google Scholar] [CrossRef]
- Li, L. Survival prediction of diffuse large-B-cell lymphoma based on both clinical and gene expression information. Bioinformatics 2006, 22, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Pineda, S.; Real, F.X.; Kogevinas, M.; Carrato, A.; Chanock, S.J.; Malats, N.; Van Steen, K. Integration Analysis of Three Omics Data Using Penalized Regression Methods: An Application to Bladder Cancer. PLoS Genet. 2015, 11, e1005689. [Google Scholar] [CrossRef] [PubMed]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Goodison, S.; Li, J.; Liu, L.; Farmerie, W. Improved breast cancer prognosis through the combination of clinical and genetic markers. Bioinformatics 2007, 23, 30–37. [Google Scholar] [CrossRef]
- Bernal Rubio, Y.L.; González Reymúndez, A.; Wu, K.-H.H.; Griguer, C.E.; Steibel, J.P.; de Los Campos, G.; Doseff, A.; Gallo, K.; Vazquez, A.I. Whole-Genome Multi-omic Study of Survival in Patients with Glioblastoma Multiforme. G3 (Bethesda) 2018, 8, 3627–3636. [Google Scholar] [CrossRef] [PubMed]
- Boulesteix, A.L.; De Bin, R.; Jiang, X.; Fuchs, M. IPF-LASSO: Integrative L1-Penalized Regression with Penalty Factors for Prediction Based on Multi-Omics Data. Comput. Math. Methods Med. 2017, 2017, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep learning–based multi-omics integration robustly predicts survival in liver cancer. Clin. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Garali, I.; Adanyeguh, I.M.; Ichou, F.; Perlbarg, V.; Seyer, A.; Colsch, B.; Moszer, I.; Guillemot, V.; Durr, A.; Mochel, F.; et al. A strategy for multimodal data integration: Application to biomarkers identification in spinocerebellar ataxia. Brief. Bioinform. 2018, 19, 1356–1369. [Google Scholar] [CrossRef] [PubMed]
- González-Reymúndez, A.; De Los Campos, G.; Gutiérrez, L.; Lunt, S.Y.; Vazquez, A.I. Prediction of years of life after diagnosis of breast cancer using omics and omic-by-treatment interactions. Eur. J. Hum. Genet. 2017, 25, 538–544. [Google Scholar] [CrossRef]
- Jayawardana, K.; Schramm, S.J.; Haydu, L.; Thompson, J.F.; Scolyer, R.A.; Mann, G.J.; Müller, S.; Yang, J.Y.H. Determination of prognosis in metastatic melanoma through integration of clinico-pathologic, mutation, mRNA, microRNA, and protein information. Int. J. Cancer 2015, 136, 863–874. [Google Scholar] [CrossRef]
- De Maturana, E.L.; Chanok, S.J.; Picornell, A.C.; Rothman, N.; Herranz, J.; Calle, M.L.; García-Closas, M.; Marenne, G.; Brand, A.; Tardón, A.; et al. Whole Genome Prediction of Bladder Cancer Risk With the Bayesian LASSO. Genet. Epidemiol. 2014, 38, 467–476. [Google Scholar] [CrossRef]
- Seoane, J.A.; Day, I.N.M.; Gaunt, T.R.; Campbell, C. A pathway-based data integration framework for prediction of disease progression. Bioinformatics 2014, 30, 838–845. [Google Scholar] [CrossRef]
- Thompson, J.A.; Marsit, C.J. A Methylation-To-Expression Feature Model for Generating Accurate Prognostic Risk Scores and Identifying Disease Targets in Clear Cell Kidney Cancer. Pacific Symp. Biocomput. 2017, 2017, 509–520. [Google Scholar] [CrossRef]
- Thompson, J.A.; Christensen, B.C.; Marsit, C.J. Methylation-to-Expression Feature Models of Breast Cancer Accurately Predict Overall Survival, Distant-Recurrence Free Survival, and Pathologic Complete Response in Multiple Cohorts. Sci. Rep. 2018, 8, 5190. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, M.H.; Horlings, H.M.; van de Vijver, M.J.; Reinders, M.J.T.; Wessels, L.F.A. Integration of clinical and gene expression data has a synergetic effect on predicting breast cancer outcome. PLoS ONE 2012, 7, e40358. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.I.; de los Campos, G.; Klimentidis, Y.C.; Rosa, G.J.M.; Gianola, D.; Yi, N.; Allison, D.B. A comprehensive genetic approach for improving prediction of skin cancer risk in humans. Genetics 2012, 192, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Song, N.; Shen, R.; Arora, A.; Machiela, M.J.; Song, L.; Landi, M.T.; Ghosh, D.; Chatterjee, N.; Baladandayuthapani, V.; et al. Integrating Clinical and Multiple Omics Data for Prognostic Assessment across Human Cancers. Sci. Rep. 2017, 7, 16954. [Google Scholar] [CrossRef]
- Abraham, G.; Inouye, M. Genomic risk prediction of complex human disease and its clinical application. Curr. Opin. Genet. Dev. 2015, 30, 10–16. [Google Scholar] [CrossRef]
- Shen, R.; Mo, Q.; Schultz, N.; Seshan, V.E.; Olshen, A.B.; Huse, J.; Ladanyi, M.; Sander, C. Integrative subtype discovery in glioblastoma using iCluster. PLoS ONE 2012. [Google Scholar] [CrossRef]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef] [PubMed]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Brief. Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, F.; Szolovits, P. Tensor factorization toward precision medicine. Brief. Bioinform. 2017, 18, 511–514. [Google Scholar] [CrossRef]
- López de Maturana, E.; Pineda, S.; Brand, A.; Van Steen, K.; Malats, N. Toward the integration of Omics data in epidemiological studies: still a “long and winding road”. Genet. Epidemiol. 2016, 40, 558–569. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Reference | Title | Outcome | Big Data: Omics and Image Data | Non-Omics | Objective | Model Performance | Approach |
|---|---|---|---|---|---|---|---|
| [25] | Classification based on extensions of LS-PLS using logistic regression: application to clinical and multiple genomic data | Dataset1: Response of childhood malignant embryonal tumors of the CNS to therapy Dataset2: ER status in breast cancer | Dataset1: gene expression data Dataset2: somatic CNA | Dataset1: sex, age, chemo CX, chemo VP. Dataset2: grade, tumor stage, HER2 status, tumor size, progesterone receptor status | Dataset1: Prediction performance: AUC Misclassification rates Dataset2: predict ER stratification of a novel breast tumor to select appropriate treatment: AUC Misclassification rates | Dataset1: AUCnon-omics = 0.60 AUComics = 0.92 AUCOnO = 0.82−0.90 Dataset2: AUCnon-omics = 0.87 AUComics = 0.84 AUCOnO = 0.93 | Conditional modeling |
| [34] | Whole-genome multi-omic study of survival in patients with glioblastoma multiform | Survival time in glioblastoma | TCGA: SNP + methylation + CNV + gene expression | TCGA: Sex + use of temozolomide | Predictive ability: AUC | AUCnon-omics = 0.71 AUComics = non-provided AUCOnO = 0.72 | Joint modeling |
| [24] | Survival prediction from clinico-genomic models—a comparative study | BrCa dataset: Survival time DLBCL dataset: Survival time Neuroblastoma dataset: Survival time | BrCa dataset: gene expression DLBCL dataset: gene expression Neuroblastoma dataset: microarray gene expression | BrCa dataset: tumor diameter, lymph node status, grade and NPI DLBCL dataset: International Prognostic Index (IPI) Neuroblastoma dataset: NB2004 stratification index | Prediction performance: deviance | Breast cancer dataset: DiDnon-omics = −12.5 DiDomics = −14 to −2 DiDOnO = −14 to −10 DLBCL dataset: DiDnon-omics = −12.5 DiDomics = 0 to −9 DiDOnO =−8 to−19 Neuroblastoma dataset: DiDnon-omics = −42 DiDomics = −28 to −45 DiDOnO =−40 to −50 | Conditional and joint modeling |
| [35] | IPF-LASSO: Integrative L1-penalized regression with penalty factors for prediction based on multi-omics data | AML dataset: OS BrCa dataset: Distant relapse free survival time Pathological response (binary) | AML dataset (TCGA): microarray gene expression somatic CNA BrCa dataset: microarray gene expression | AML dataset (TCGA): age, % blast cells in bone marrow, white blood cell count per mm3, and sex BrCa dataset: age, nodal status, tumor size, grade, estrogen receptor, and progesterone receptor | Predictive ability: Prediction error curves, Brier score, integrated Brier score | AML dataset: IBSnon-omics = non-provided IBSomics = non-provided IBSOnO = 0.211–0.196 Breast cancer dataset: IBSnon-omics = non-provided IBSomics = non-provided IBSOnO = 0.134–0.127 | Joint modeling |
| [36] | Deep learning based multi-omics integration robustly predicts survival in liver cancer | TCGA dataset: Survival LIRI-JP cohort: Survival NCI cohort: Survival Chinese cohort: Survival E-TABM-36 cohort: Survival Hawaiian cohort: Survival | TCGA dataset: RNA-seq, miRNA-seq, DNA methylation LIRI-JP cohort: RNA-seq NCI cohort: microarray gene expression Chinese cohort: miRNA E-TABM-36 cohort: gene expression Hawaiian cohort: DNA methylation | TCGA dataset: Stage, grade, race, gender, age, and risk factor | Predictive ability: C-index, Brier score Long-rank p-value | LIRI-JPa: C-indexnon-omics = 0.55 C-indexomics = 0.75 C-indexOnO = 0.74 NCI a: C-indexnon-omics = 0.45 C-indexomics = 0.67 C-indexOnO = 0.65 | Independent modeling |
| [37] | A strategy for multimodal data integration: application to biomarkers identification in spinocerebellar ataxia | SCA dataset: SCA subtypes and controls | SCA dataset: 754 metabolites | SCA dataset: MRS of the cerebellum, calorimetry information, volume of the pons | Graphical Reliability of parameter estimates % of times a variable has a non-null weight | Non-provided | Joint modeling |
| [38] | Prediction of years of life after diagnosis of breast cancer using omics and omic-by-treatment interactions | METABRIC dataset: Log (survival time) | METABRIC dataset: CNV, gene expression (GE) Also interactions with treatment: GExCT, GExRT, GExHT | METABRIC dataset: Age, cancer subtype, histological type, Nottingham Prognostic Index (tumor size, grade and nodal involvement), treatment | Prediction accuracy: AUC % of variance explained Definition of two groups: high and low risk | AUCnon-omics = 0.72–0.77 AUComics = non-provided AUCOnO = 0.74–0.81 b | Joint modeling |
| [39] | Determination of prognosis in metastatic melanoma through integration of clinic-pathologic, mutation, mRNA, microRNA, and protein information | MIA dataset: Survival grouped into good prognosis and poor prognosis | MIA dataset: mRNA, somatic mutation, microRNA expression, protein expression | MIA dataset: Nineteen clinical/pathological data: ulceration and thickness of primary tumor, number of lymph nodes with metastases, and size of nodal metastasis at the time of staging and others | Prediction error rate (ER) Kaplan-Meier curves | ERnon-omics = 30% ERmRNA = 25% ERprotein = 35% ERmicroRNA = 37% EROnO = 29–33% | Independent modeling |
| [40] | Whole Genome Prediction of Bladder Cancer Risk With the Bayesian LASSO | SBC/EPICURO dataset: BC risk | EPICURO/SBC dataset: SNP | SBC/EPICURO dataset: age + gender + region + smoking | Prediction: AUC | AUCnon-omics = 0.65 AUComics = 0.53 AUCOnO = 0.65 | Joint modeling |
| [20] | Prediction of non-muscle invasive bladder cancer outcomes assessed by innovative multimarker prognostic models | SBC/EPICURO dataset: Time to first recurrence (TFR) Time to progression (TP) | EPICURO/SBC dataset: SNP | SBC/EPICURO dataset: TFR: Area + gender + # of tumors + TSG + tumor size + treatment TP: Area + age + # of tumors + TSG + # of recurrences + treatment | Prediction: AUC, R2 | TFR c: AUCnon-omics = 0.62 AUComics = 0.55 AUCOnO = 0.61 | Joint modeling |
| [41] | A pathway based data integration framework for prediction of disease progression | METABRIC dataset: Survival vs. not survival at 2000 days | METABRIC dataset: Gene expression, CNV | METABRIC dataset: ER status only, disease &treatment group, grade of disease, stage, histological type, HER2 status, age, tumor size, NPI (tumor size, lymph node, grade), tumor cellularity, PAM50-based subtype | Accuracy | Accnon-omics = non-provided Accomics = 0.64–0.71 AccOnO = 0.66–0.80 | Joint modeling |
| [42] | A methylation-to-expression feature model for generating accurate prognostic risk scores and identifying disease targets in clear cell kidney cancer | TCGA dataset: OS in clear cell renal cell carcinoma | TCGA dataset: Gene expression (RNA seq), DNA methylation profile | TCGA dataset: Age, sex, tumor stage | Classification performance: C-index | C-indexnon-omics = 0.776 C-indexomics = 0.702 C-indexOnO = 0.792 | Independent modeling approach |
| [43] | Methylation-to-expression feature models of breast cancer accurately predict overall survival, distant-recurrence free survival, and pathologic complete response in multiple cohorts | TCGA dataset: OS (time) Terunuma dataset: OS (time) Kao dataset: OS (time) Hatzis1 dataset: Distant recurrence-free survival (time) Hatzis2 dataset: Distant recurrence-free survival (time) Pathologic complete response in BC (binary) | TCGA dataset: gene expression and methylation profiles Terunuma dataset: gene expression Kao dataset: gene expression Hatzis1 dataset: gene expression Hatzis2 dataset: gene expression | All datasets: AJCC stage, Age, ER status, PR status, HER2 status, PAM50-based subtype | Classification performance: C-index, AUC | OS TCGA d: C-indexnon-omics = 0.75 C-indexomics e = 0.69 C-indexOnO = 0.79 | Independent modeling |
| [44] | Integration of Clinical and Gene Expression Data Has a Synergetic Effect on Predicting Breast Cancer Outcome | Vijver dataset: Poor/good outcome Other datasets: Poor/good outcome | Vijver dataset: Expression data Other datasets: Expression data | Vijver dataset: 45 clinical variables Otherdatasets: age, tumor size, grade, ER stats, lymph node, NPI | Classification performance: error rate, AUC | Vijver dataset: AUCnon-omics = 0.75 AUComics = 0.74 AUCOnO = 0.74-0.78 | Independent and Joint modeling |
| [45] | A Comprehensive Genetic Approach for Improving Prediction of Skin Cancer Risk in Humans | Kreger’s dataset: Skin cancer risk | Kreger’s dataset: SNPs | Kreger’s dataset: Age, ethnicity | Classification performance: AUC | AUCnon-omics = 0.53–0.54 AUComics = 0.63–0.64 AUCOnO = non-provided | Joint modeling |
| [46] | Integrating Clinical and Multiple Omics Data for Prognostic Assessment across Human Cancers | TCGA datasets: Survival prediction in multiple cancer types | TCGA datasets: Significant SNPs (PRS, as fixed effect), somatic mutation, mRNA, miRNA, methylation, copy number, immune and metagenes signatures | TCGA datasets: Age, stage, Lauren classification (STAD) Also depends on the cancer type MammaPrint and PAM50 gene signatures | Classification performance: C-index | Ovarian and HNSC f: C-indexnon-omics = 0.60; 0.61 C-indexomics = 0.61; 0.61 C-indexOnO = 0.64;0.64 | Independent modeling |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López de Maturana, E.; Alonso, L.; Alarcón, P.; Martín-Antoniano, I.A.; Pineda, S.; Piorno, L.; Calle, M.L.; Malats, N. Challenges in the Integration of Omics and Non-Omics Data. Genes 2019, 10, 238. https://doi.org/10.3390/genes10030238
López de Maturana E, Alonso L, Alarcón P, Martín-Antoniano IA, Pineda S, Piorno L, Calle ML, Malats N. Challenges in the Integration of Omics and Non-Omics Data. Genes. 2019; 10(3):238. https://doi.org/10.3390/genes10030238
Chicago/Turabian StyleLópez de Maturana, Evangelina, Lola Alonso, Pablo Alarcón, Isabel Adoración Martín-Antoniano, Silvia Pineda, Lucas Piorno, M. Luz Calle, and Núria Malats. 2019. "Challenges in the Integration of Omics and Non-Omics Data" Genes 10, no. 3: 238. https://doi.org/10.3390/genes10030238