Pituitary Transcriptomic Study Reveals the Differential Regulation of lncRNAs and mRNAs Related to Prolificacy in Different FecB Genotyping Sheep

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. RNA Extraction and Library Preparation
2.3. Clustering, Sequencing and Transcriptome Assembly
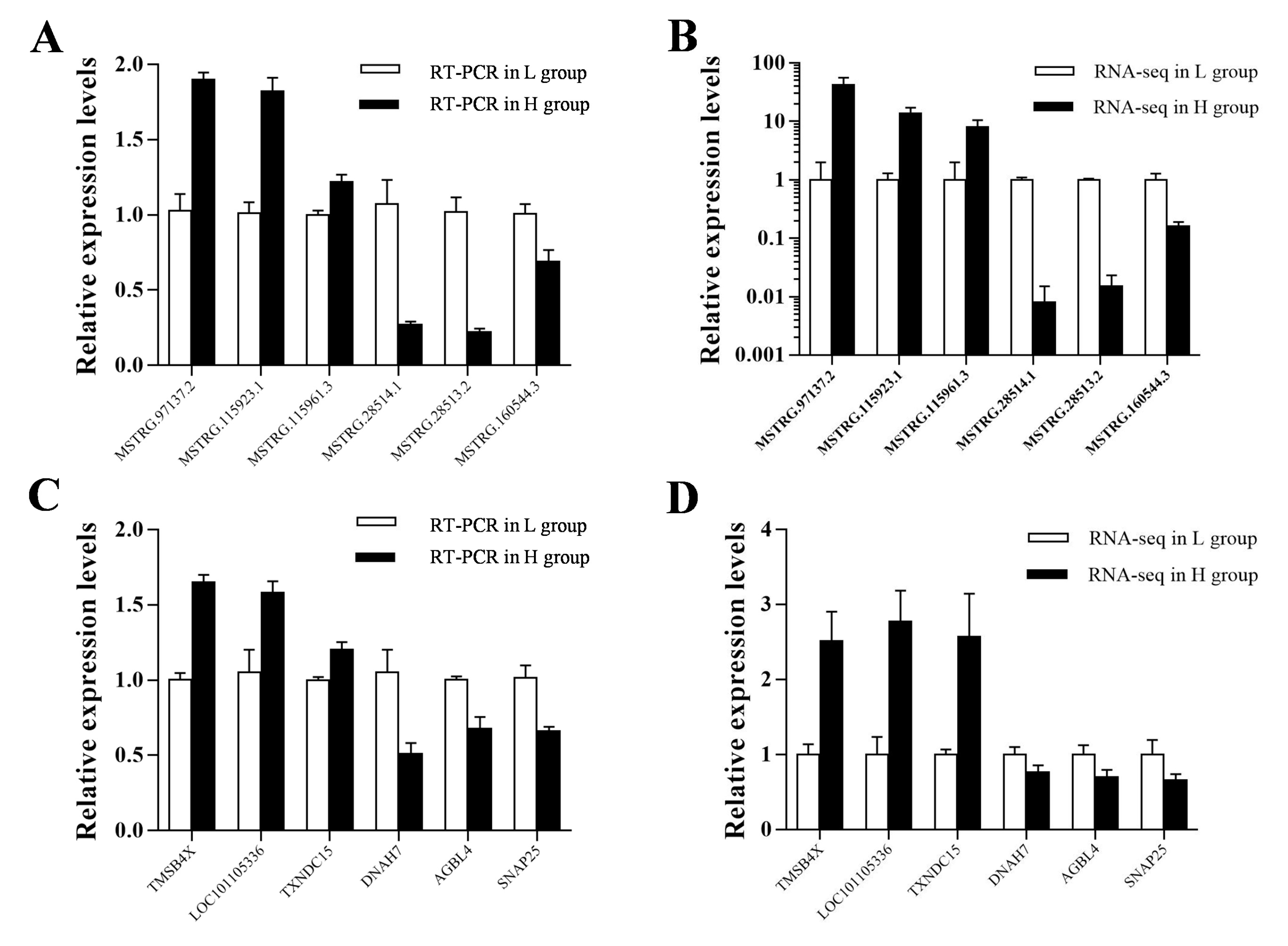
2.4. Differential Expression Analysis and qRT-PCR Validation
2.5. Target Gene Prediction and Functional Annotation Analysis
2.6. Construction of the LncRNA-gene Co-Expression Network
2.7. Pituitary Cells Isolation, Transfection and qRT-PCR Validation
2.8. Immunofluorescence Staining of Primary Pituitary Cells
2.9. Statistical Analysis
3. Results
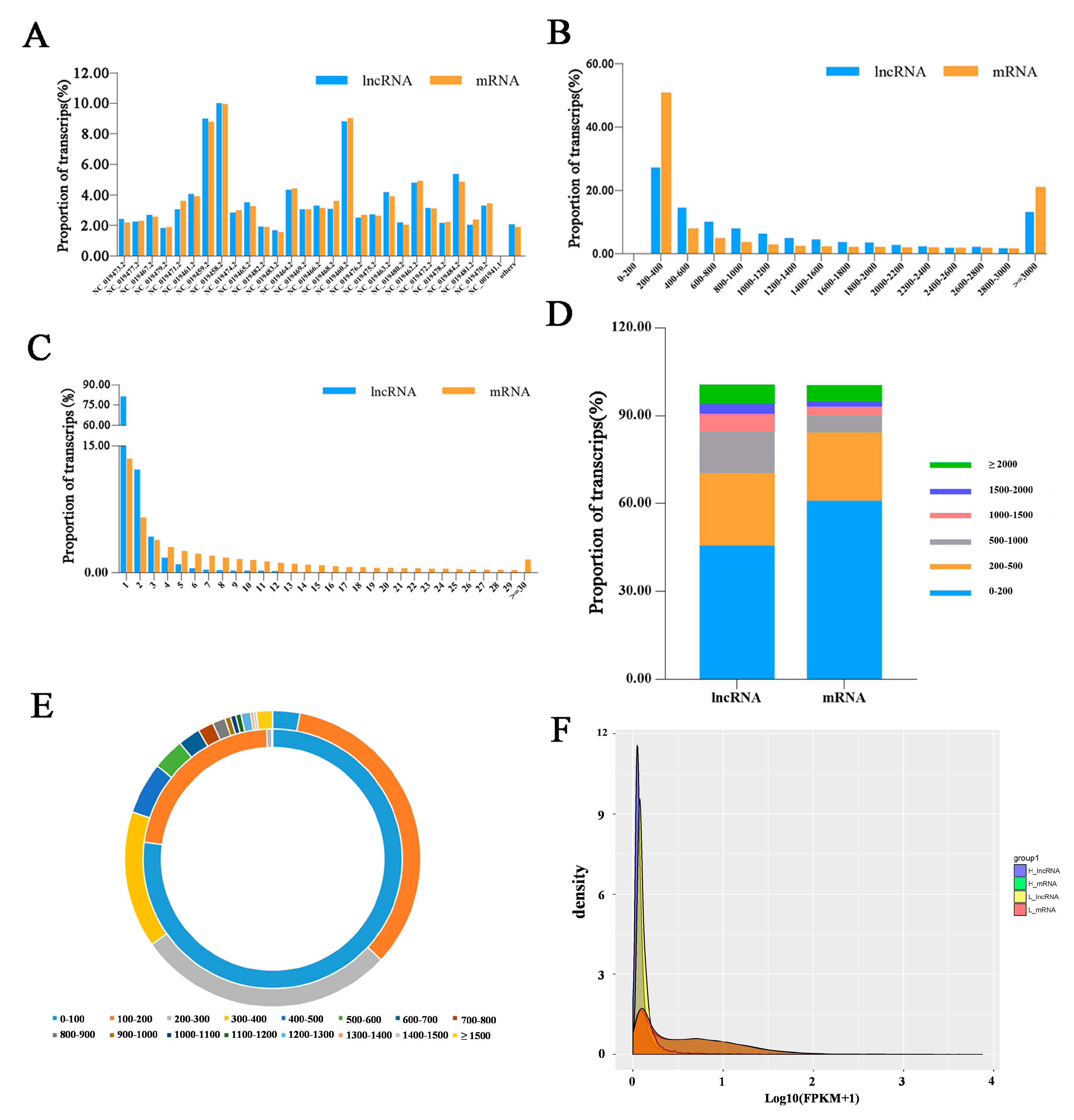
3.1. Overview of Sequencing Data in Sheep Pituitary Tissue
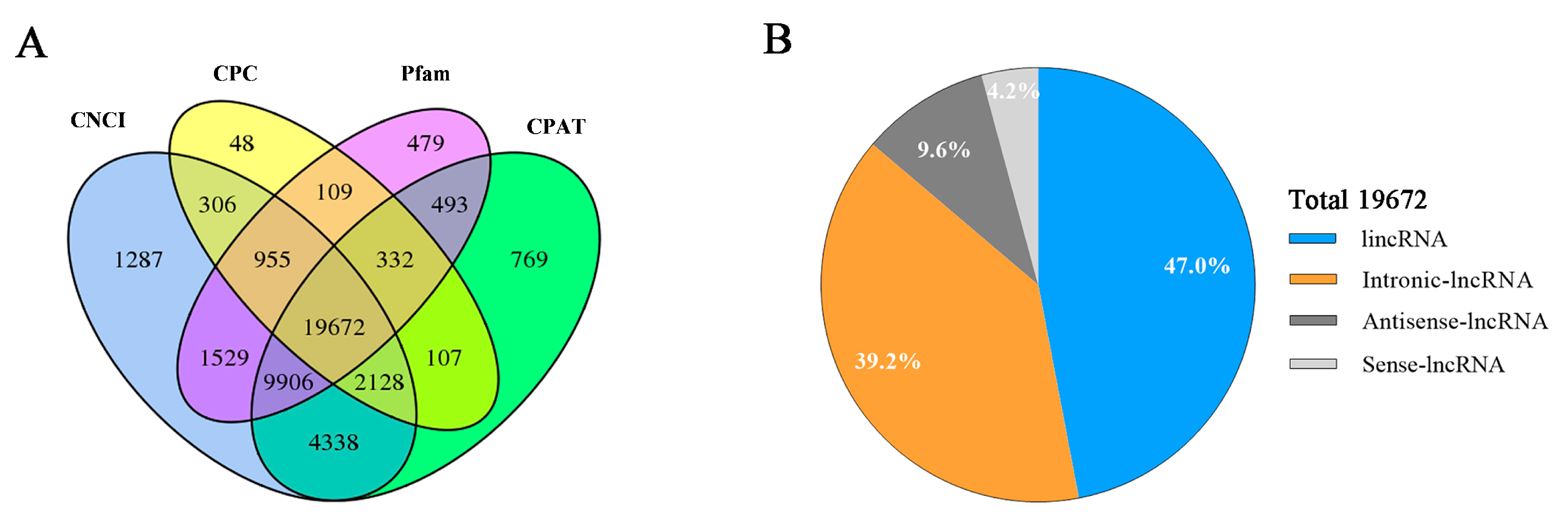
3.2. Identification of LncRNAs and mRNAs in Hu Sheep Pituitary Tissue
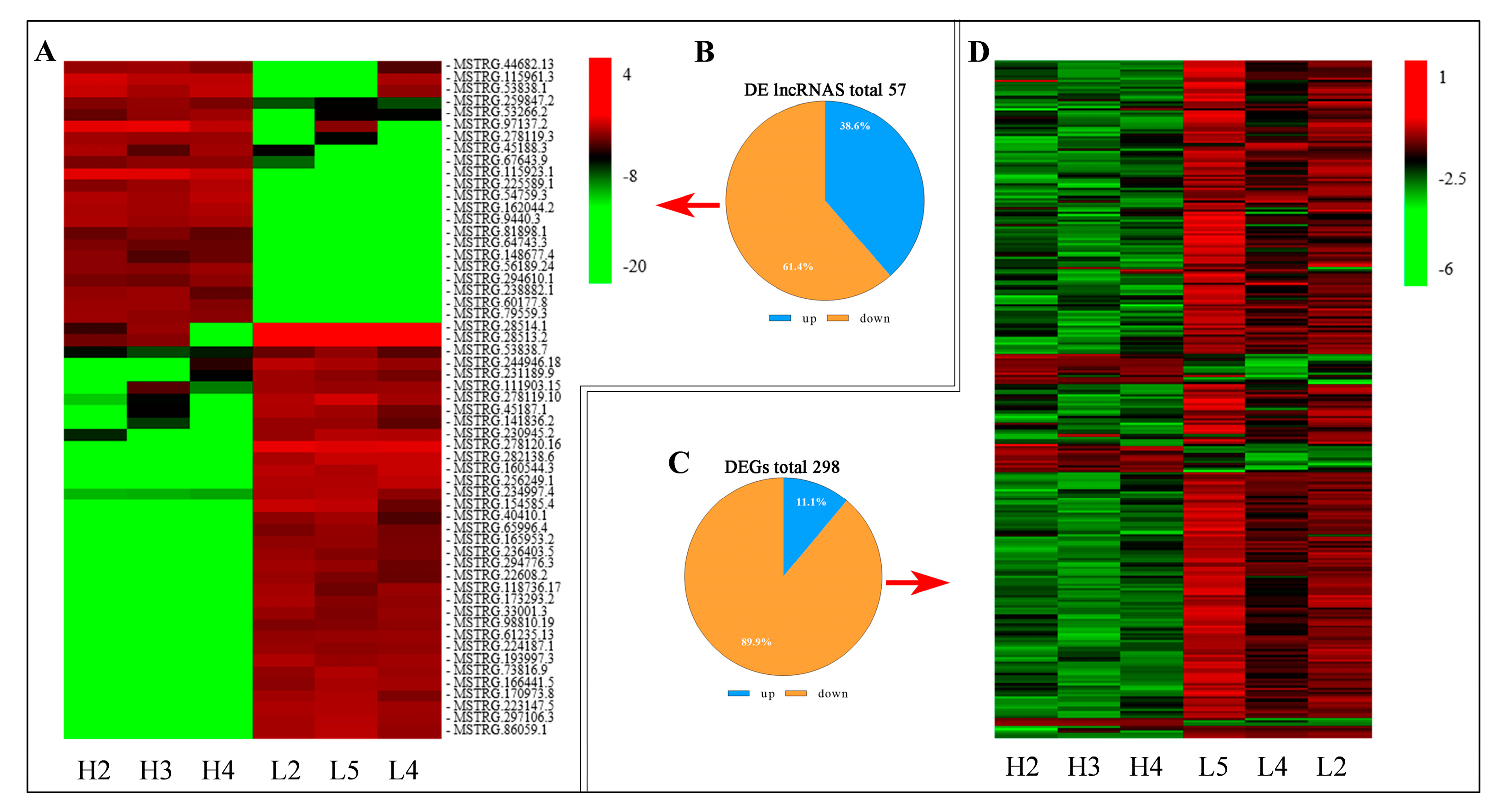
3.3. The Profiling and Verification of DE LncRNA and DEGs of Sheep Pituitary
3.4. GO and KEGG Analysis of DEGs
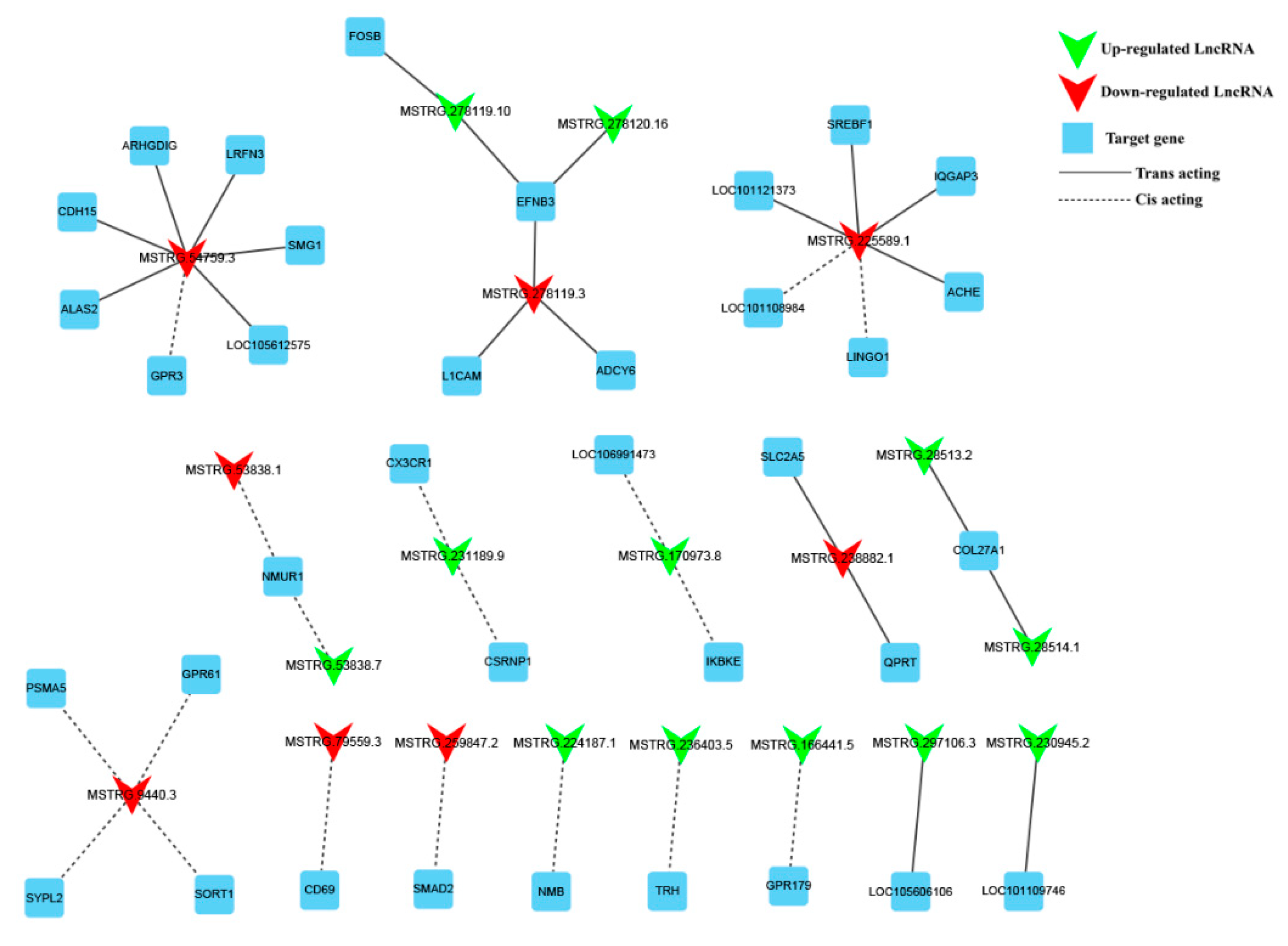
3.5. Screening of Potential Functional LncRNAs Involved in Hu Sheep Reproduction
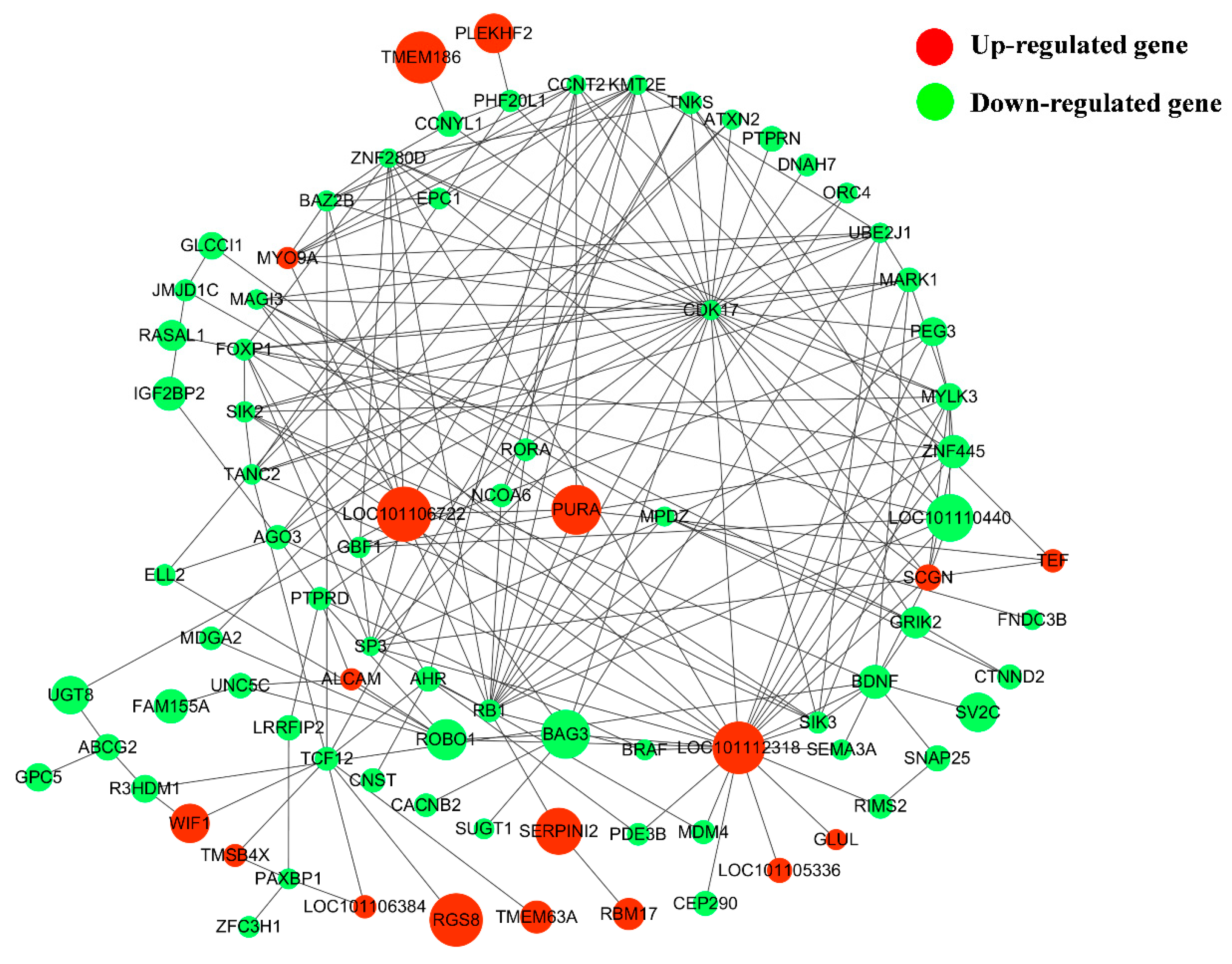
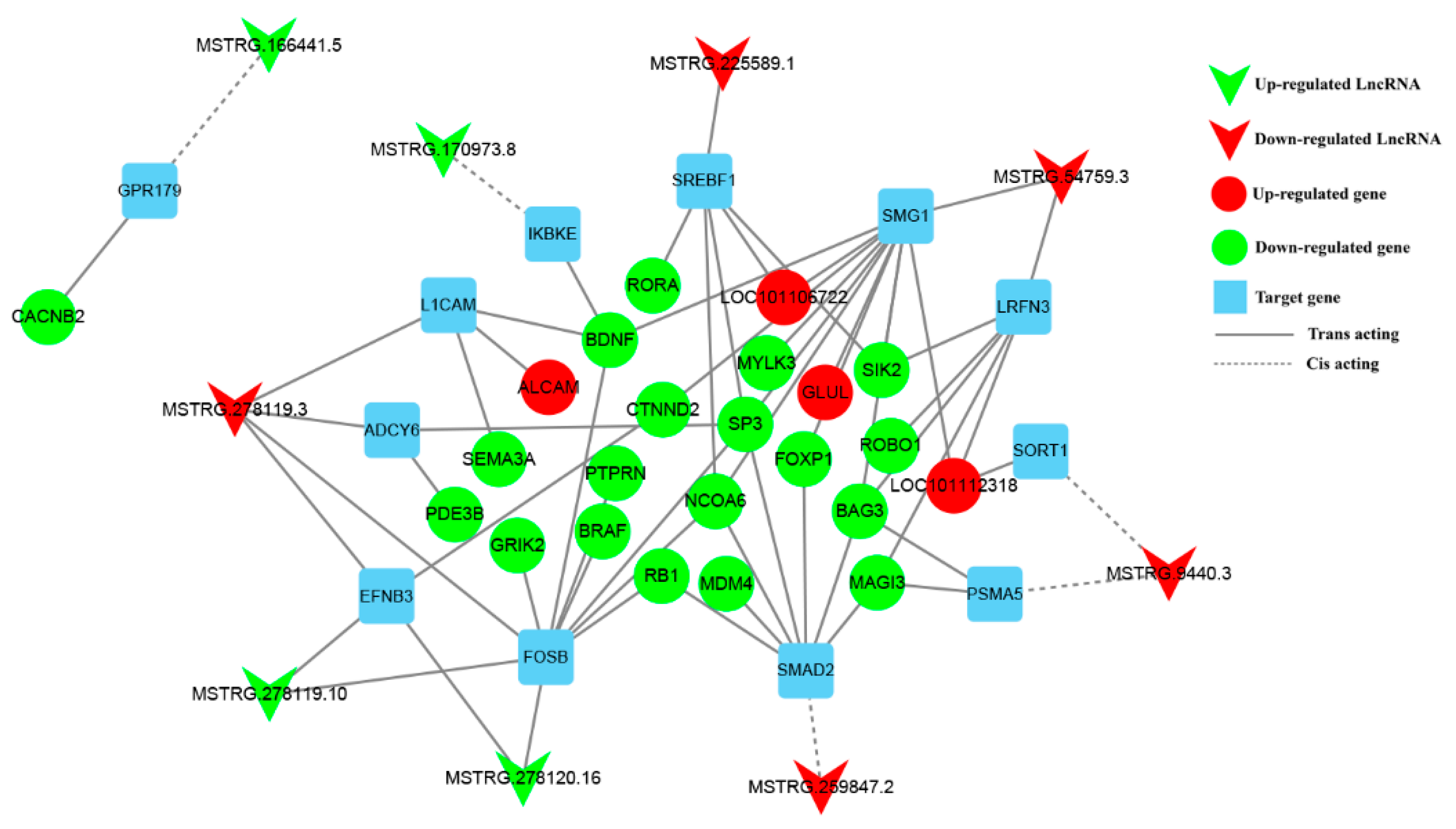
3.6. Construction of DE lncRNA-Target Gene-DEG Regulated Networks
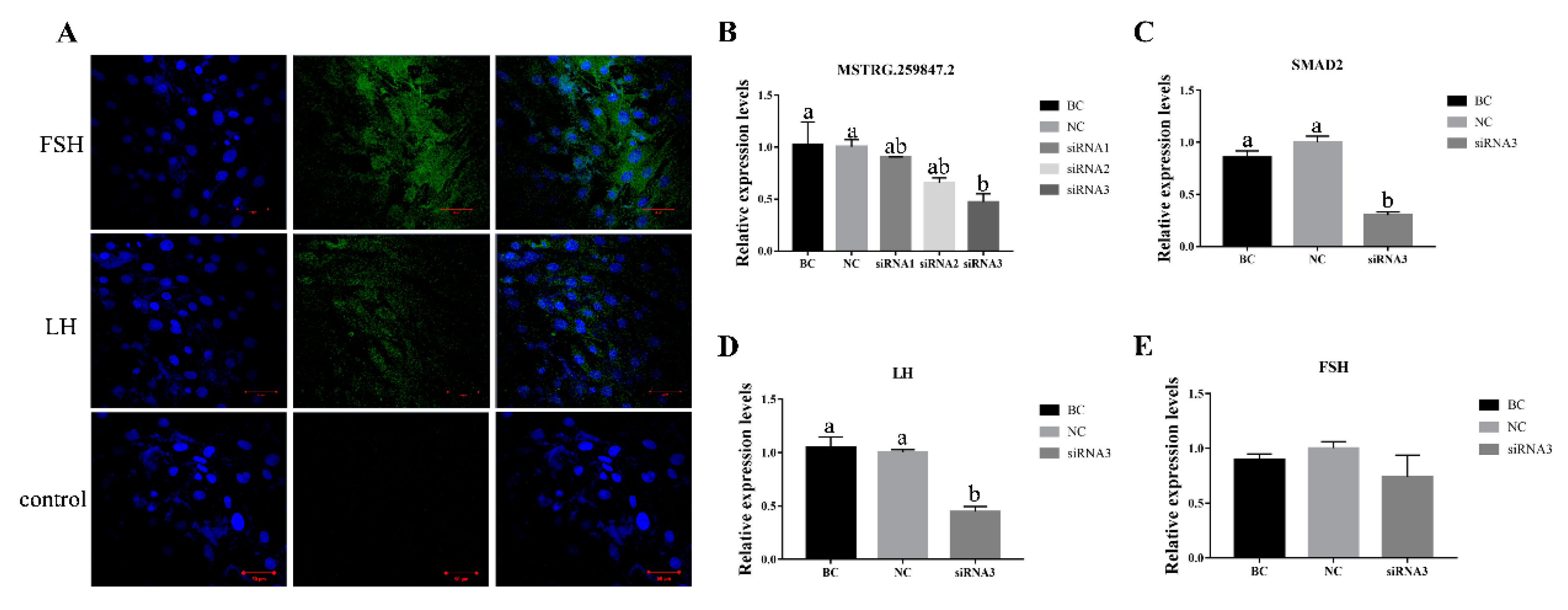
3.7. Verification of MSTRG.259847.2 and Its Target Gene SMAD2 in Sheep Primary Pituitary Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mulsant, P.; Lecerf, F.; Fabre, S.; Schibler, L.; Monget, P.; Lanneluc, I.; Pisselet, C.; Riquet, J.; Monniaux, D.; Callebaut, I. Mutation in Bone Morphogenetic Protein Receptor-IB is Associated with Increased Ovulation Rate in Booroola Mérino Ewes. Proc. Natl. Acad. Sci. USA 2001, 98, 5104–5109. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.; Wu, X.Y.; Juengel, J.L.; Ross, I.K.; Lumsden, J.M.; Lord, E.A.; Dodds, K.G.; Walling, G.A.; Mcewan, J.C.; O’Connell, A.R. Highly Prolific Booroola Sheep Have a Mutation in the Intracellular Kinase Domain of Bone Morphogenetic Protein IB Receptor (ALK-6) That Is Expressed in Both Oocytes and Granulosa Cells1. Biol. Reprod. 2001, 64, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.J.; Macdougall, C.; Macdougall, C.; Campbell, B.K.; Mcneilly, A.S.; Baird, D.T. The Booroola (FecB) phenotype is associated with a mutation in the bone morphogenetic receptor type 1 B (BMPR1B) gene. J. Endocrinol. 2001, 169, 1–6. [Google Scholar] [CrossRef]
- Davis, G.H.; Balakrishnan, L.; Ross, I.K.; Wilson, T.; Galloway, S.M.; Lumsden, B.M.; Hanrahan, J.P.; Mullen, M.; Mao, X.Z.; Wang, G.L. Investigation of the Booroola (FecB) and Inverdale (FecX(I)) mutations in 21 prolific breeds and strains of sheep sampled in 13 countries. Anim. Reprod. Sci. 2006, 92, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.X.; Liu, Z.H.; Jiao, C.L.; He, Y.Q.; Fang, L.; Ye, S.C.; Chen, G.H.; Wang, J.Y. Mutations in BMPR-IB and BMP-15 genes are associated with litter size in Small Tailed Han sheep (Ovis aries). J. Anim. Sci. 2007, 85, 598. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, N.M. A review of the effects of the Booroola gene (FecB) on sheep production. Small Rumin. Res. 2009, 85, 75–84. [Google Scholar] [CrossRef]
- Wang, W.M.; Liu, S.J.; Li, F.D.; Pan, X.Y.; Li, C.; Zhang, X.X.; Ma, Y.J.; La, Y.F.; Xi, R.; Li, T.F. Polymorphisms of the Ovine BMPR-IB, BMP-15 and FSHR and Their Associations with Litter Size in Two Chinese Indigenous Sheep Breeds. Int. J. Mol. Sci. 2015, 16, 11385–11397. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Hombach, S.; Kretz, M. Non-coding RNAs: Classification, Biology and Functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Panda, A.; Kang, M.J.; Xu, J.; Selimyan, R.; Yoon, J.H.; Martindale, J.L.; De, S.; Wood, W.H., 3rd; Becker, K.G.; et al. Senescence-associated lncRNAs: Senescence-associated long noncoding RNAs. Aging Cell 2013, 12, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Arun, G.; Akhade, V.S.; Donakonda, S. mrhl RNA, a Long Noncoding RNA, Negatively Regulates Wnt Signaling through Its Protein Partner Ddx5/p68 in Mouse Spermatogonial Cells. Mol. Cell. Biol. 2012, 32, 3140. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S.; Das, S.; Chakrabarti, J. Long noncoding RNAs: New players in the molecular mechanism for maintenance and differentiation of pluripotent stem cells. Stem Cells Dev. 2013, 22, 2240–2253. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, B.B.; Olcese, U.; Cabrera, J.R.; Horabin, J.I. An interactive network of long non-coding RNAs facilitates the Drosophila sex determination decision. Biochim. Biophys. Acta 2014, 1839, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Notani, D.; Ma, Q.; Tanasa, B.; Nunez, E.; Chen, A.Y.; Merkurjev, D.; Zhang, J.; Ohgi, K.; Song, X.; et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 2013, 498, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Mau, M.; Corral, J.M.; Vogel, H.; Melzer, M.; Fuchs, J.; Kuhlmann, M.; de Storme, N.; Geelen, D.; Sharbel, T.F. The Conserved Chimeric Transcript UPGRADE2 Is Associated with Unreduced Pollen Formation and Is Exclusively Found in Apomictic Boechera Species. Plant. Physiol. 2013, 163, 1640–1659. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.L.; Liu, M.; Yang, Y.Y.; Yang, H.X.; Liao, Q.P.; Bai, Y.; Li, Y.X.; Li, D.; Peng, C.; Wang, Y.L. The imprinted H19 gene regulates human placental trophoblast cell proliferation via encoding miR-675 that targets Nodal Modulator 1 (NOMO1). RNA Biol. 2012, 9, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Keniry, A.; Oxley, D.; Monnier, P.; Kyba, M.; Dandolo, L.; Smits, G.; Reik, W. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat. Cell Biol. 2012, 14, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, T.; Mano, A.; Shibasaki, T. Increased expression of miR-325-3p by urocortin 2 and its involvement in stress-induced suppression of LH secretion in rat pituitary. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E781–E787. [Google Scholar] [CrossRef] [PubMed]
- Schang, A.L.; Granger, A.; Querat, B.; Bleux, C.; Cohen-Tannoudji, J.; Laverriere, J.N. GATA2-induced silencing and LIM-homeodomain protein-induced activation are mediated by a bi-functional response element in the rat GnRH receptor gene. Mol. Endocrinol. 2013, 27, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Ueharu, H.; Higuchi, M.; Nishimura, N.; Yoshida, S.; Shibuya, S.; Sensui, K.; Kato, T.; Kato, Y. Expression of Kruppel-like factor 6, KLF6, in rat pituitary stem/progenitor cells and its regulation of the PRRX2 gene. J. Reprod. Dev. 2014, 60, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Han, D.X.; Dai, L.S.; Gao, Y.; Ding, Y.; Yu, X.F.; Chen, J.; Jiang, H.; Chen, C.Z.; Zhang, J.B. A comprehensive expression profile of micrornas in rat’s pituitary. Int. J. Clin. Exp. Med. 2015, 8, 13289–13295. [Google Scholar] [PubMed]
- Han, D.X.; Sun, X.L.; Fu, Y.; Wang, C.J.; Liu, J.B.; Jiang, H.; Gao, Y.; Chen, C.Z.; Yuan, B.; Zhang, J.B. Identification of long non-coding RNAs in the immature and mature rat anterior pituitary. Sci. Rep. 2017, 7, 17780. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.L.; Cao, S.W.; Ou, Q.S.; Yang, B.; Zheng, S.H.; Tang, J.; Chen, J.; Hu, Y.W.; Zheng, L.; Wang, Q. The long non-coding RNA PTTG3P promotes cell growth and metastasis via up-regulating PTTG1 and activating PI3K/AKT signaling in hepatocellular carcinoma. Mol. Cancer 2018, 17, 93. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Zhang, Y.; Cui, H. Long noncoding RNA CCAT2 is activated by E2F1 and exerts oncogenic properties by interacting with PTTG1 in pituitary adenomas. Am. J. Cancer Res. 2018, 8, 245–255. [Google Scholar] [PubMed]
- Leka-Emiri, S.; Chrousos, G.P.; Kanaka-Gantenbein, C. The mystery of puberty initiation: Genetics and epigenetics of idiopathic central precocious puberty (ICPP). J. Endocrinol. Investig. 2017, 40, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.T.; Bu, D.C.; Zhao, G.G.; Yu, K.T.; Zhang, C.H.; Liu, Y.N.; Chen, R.S.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.Q.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.L.; Yang, H.; Zhang, Y.L.; Ren, C.F.; Nie, H.T.; Fan, Y.X.; Zhou, W.J.; Wang, S.T.; Feng, X.; Wang, F. Characterization of GALNTL5 gene sequence and expression in ovine testes and sperm. Theriogenology 2017, 95, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, G.; Turco, V.L.; Potorti, A.G.; Rando, R.; Licata, P.; Dugo, G. Statistical analysis of heavy metals in Cerastoderma edule glaucum and Venerupis aurea laeta from Ganzirri Lake, Messina (Italy). Environ. Monit. Assess 2013, 185, 7517–7525. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Li, F.; Wang, F.; Zhang, G.; Pang, J.; Ren, C.; Zhang, T.; Yang, H.; Wang, Z.; Zhang, Y. Genome-wide differential expression profiling of mRNAs and lncRNAs associated with prolificacy in Hu sheep. Biosci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Luo, Q.; Zhao, H.; Qin, X. Co-expression analysis and identification of fecundity-related long non-coding RNAs in sheep ovaries. Sci. Rep. 2016, 6, 39398. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.Y.; Luo, Q.M.; Zhao, H.J.; Qin, X.Y. Ovarian transcriptomic study reveals the differential regulation of miRNAs and lncRNAs related to fecundity in different sheep. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Qin, Q.L. Genome-wide transcriptome analysis of mRNAs and microRNAs in Dorset and Small Tail Han sheep to explore the regulation of fecundity. Mol. Cell Endocrinol. 2015, 402, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, M.; Nanekarani, S.; Hosseini, S.D. Mutation in BMPR-IB gene is associated with litter size in Iranian Kalehkoohi sheep. Anim. Reprod. Sci. 2014, 147, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Sive, H.; Bartel, D.P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 2011, 147, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.W.; Bartel, D.P. Long noncoding RNAs in C. elegans. Genome Res. 2012, 22, 2529–2540. [Google Scholar] [CrossRef] [PubMed]
- Vallot, C.; Huret, C.; Lesecque, Y.; Resch, A.; Oudrhiri, N.; Bennaceur-Griscelli, A.; Duret, L.; Rougeulle, C. XACT, a long noncoding transcript coating the active X chromosome in human pluripotent cells. Nat. Genet. 2013, 45, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Appeltant, R.; Beek, J.; Vandenberghe, L.; Maes, D.; Van Soom, A. Increasing the cAMP concentration during in vitro maturation of pig oocytes improves cumulus maturation and subsequent fertilization in vitro. Theriogenology 2015, 83, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Zemkova, H.; Kucka, M.; Bjelobaba, I.; Tomic, M.; Stojilkovic, S.S. Multiple cholinergic signaling pathways in pituitary gonadotrophs. Endocrinology 2013, 154, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, T.; Otsuka, F.; Suzuki, J.; Takeda, M.; Inagaki, K.; Kano, Y.; Otani, H.; Mimura, Y.; Ogura, T.; Makino, H. Mutual regulation of follicle-stimulating hormone signaling and bone morphogenetic protein system in human granulosa cells. Biol. Reprod. 2006, 74, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Biran, J.; Palevitch, O.; Ben-Dor, S.; Levavi-Sivan, B. Neurokinin Bs and neurokinin B receptors in zebrafish-potential role in controlling fish reproduction. Proc. Natl. Acad. Sci. USA 2012, 109, 10269–10274. [Google Scholar] [CrossRef] [PubMed]
- Indovina, P.; Pentimalli, F.; Casini, N.; Vocca, I.; Giordano, A. RB1 dual role in proliferation and apoptosis: Cell fate control and implications for cancer therapy. Oncotarget 2015, 6, 17873–17890. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Bauzon, F.; Bi, E.; Yu, J.J.; Fu, H.; Lu, Z.; Cui, J.; Jeon, H.; Zang, X.; Ye, B.H.; et al. Substituting threonine 187 with alanine in p27Kip1 prevents pituitary tumorigenesis by two-hit loss of Rb1 and enhances humoral immunity in old age. J. Biol. Chem. 2015, 290, 5797–5809. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.G.; Wang, Q.; Jia, J.; Pincas, H.; Turgeon, J.L.; Sealfon, S.C. Growth differentiation factor 9 (GDF9) forms an incoherent feed-forward loop modulating follicle-stimulating hormone β-subunit (FSHbeta) gene expression. J. Biol. Chem. 2014, 289, 16164–16175. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ho, C.C.; Bang, E.; Rejon, C.A.; Libasci, V.; Pertchenko, P.; Hebert, T.E.; Bernard, D.J. Bone morphogenetic protein 2 stimulates noncanonical SMAD2/3 signaling via the BMP type 1A receptor in gonadotrope-like cells: Implications for FSH synthesis. Endocrinology 2014, 155, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Abdoli, R.; Zamani, P.; Mirhoseini, S.Z.; Ghavi Hossein-Zadeh, N.; Nadri, S. A review on prolificacy genes in sheep. Reprod. Domest. Anim. 2016, 51, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.S.; Hu, W.P.; Di, R.; Liu, Q.Y.; Wang, X.Y.; Zhang, X.S.; Zhang, J.L.; Chu, M.X. Expression Analysis of the Prolific Candidate Genes, BMPR1B, BMP15, and GDF9 in Small Tail Han Ewes with Three Fecundity (FecB Gene) Genotypes. Animals 2018, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, K.M.; Sukanta, S.; Bhuyan, M.A.; Ali, S.M.; Ishaque, A.M.; Hai, M.A. Plasma LH and FSH levels in azoospermia and in normal male and female human subjects. Bangladesh Med. Res. Counc. Bull. 1993, 19, 58–62. [Google Scholar] [PubMed]
- Mcnatty, K.P.; Hudson, N.L.; Lun, S.; Heath, D.A.; Shaw, L.; Condell, L.; Phillips, D.J.; Clarke, I.J. Gonadotrophin-releasing hormone and the control of ovulation rate by the FecB gene in Booroola ewes. J. Reprod. Fertil. 1993, 98, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.J.; Hudson, N.L.; Mcnatty, K.P. Effects of ovariectomy and genotype on bioactive FSH in plasma and pituitary of Booroola ewes. J. Reprod. Fertil. 1993, 98, 559. [Google Scholar] [CrossRef] [PubMed]
- Matusiak, D.; Glover, S.; Nathaniel, R.; Matkowskyj, K.; Yang, J.X.; Benya, R.V. Neuromedin B and its receptor are mitogens in both normal and malignant epithelial cells lining the colon. Am. J. Physiol.-Gastr. L 2005, 288, G718–G728. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, K.J.; Ortiga-Carvalho, T.M.; Cabanelas, A.; Veiga, M.A.L.C.; Aoki, K.; Ohki-Hamazaki, H.; Wada, K.; Wada, E.; Pazos-Moura, C.C. Disruption of neuromedin B receptor gene results in dysregulation of the pituitary-thyroid axis. J. Mol. Endocrinol. 2006, 36, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Nakamachi, T.; Inoue, K.; Ikeda, R.; Kitamura, K.; Minamino, N.; Shioda, S.; Miyata, A. Autocrine effects of neuromedin B stimulate the proliferation of rat primary osteoblasts. J. Endocrinol. 2013, 217, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kameda, H.; Miyoshi, H.; Shimizu, C.; Nagai, S.; Nakamura, A.; Kondo, T.; Chida, D.; Atsumi, T. Expression and Regulation of Neuromedin B in Pituitary Corticotrophs of Male Melanocortin 2 Receptor-Deficient Mice. Endocrinology 2014, 155, 2492–2499. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.; Mizukami, Y.; Watanabe, K.; Sakaguti, S.; Kadokawa, H. Deep sequencing of the transcriptome in the anterior pituitary of heifers before and after ovulation. J. Vet. Med. Sci. 2017, 79, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Yamamoto, A.; Okada, T.; Matsumura, E.; Nose, E.; Kogure, K.; Kojima, S.; Haga, T. Identification of surrogate ligands for orphan G protein-coupled receptors. Life Sci. 2003, 74, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, M.; Tujii, T.; Takeda, S. The N-Terminal Domain of GPR61, an Orphan G-Protein-Coupled Receptor, Is Essential for Its Constitutive Activity. J. Neurosci. Res. 2009, 87, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.L.; Steurer, M.A.; Aronstam, R.S. Constitutive Activity among Orphan Class-A G Protein Coupled Receptors. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, U.; Kadokawa, H. The nonsteroidal mycoestrogen zearalenone and its five metabolites suppress LH secretion from the bovine anterior pituitary cells via the estradiol receptor GPR30 in vitro. Theriogenology 2015, 84, 1342–1349. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, U.; Rudolf, F.O.; Pandey, K.; Kadokawa, H. The non-steroidal mycoestrogen zeranol suppresses luteinizing hormone secretion from the anterior pituitary of cattle via the estradiol receptor GPR30 in a rapid, non-genomic manner. Anim. Reprod. Sci. 2015, 156, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, F.O.; Kadokawa, H. Cytoplasmic kinases downstream of GPR30 suppress gonadotropin-releasing hormone (GnRH)-induced luteinizing hormone secretion from bovine anterior pituitary cells. J. Reprod. Dev. 2016, 62, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; Radic, Z. The Cholinesterases—From Genes to Proteins. Annu. Rev. Pharm. 1994, 34, 281–320. [Google Scholar] [CrossRef] [PubMed]
- Herman, A.P.; Krawczynska, A.; Bochenek, J.; Haziak, K.; Romanowicz, K.; Misztal, T.; Antushevich, H.; Herman, A.; Tomaszewska-Zaremba, D. The effect of rivastigmine on the LPS-induced suppression of GnRH/LH secretion during the follicular phase of the estrous cycle in ewes. Anim. Reprod. Sci. 2013, 138, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Goff, K.J.; Notter, D.R.; Vanimisetti, H.B.; Knight, J.W. Strategies for rapid rebreeding of lactating ewes in the spring. Anim. Int. J. Anim. Biosci. 2014, 8, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Sejian, V.; Maurya, V.P.; Prince, L.L.; Kumar, D.; Naqvi, S.M. Effect of FecB status on the allometric measurements and reproductive performance of Garole x Malpura ewes under hot semi-arid environment. Trop. Anim. Health Prod. 2015, 47, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.M.; Clayton, E.H.; Morgan, B.; Friend, M.A. Reproductive performance in ewes fed varying levels of cut lucerne pasture around conception. Anim. Reprod. Sci. 2015, 158, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, S.; Raju, P.; Rao, S.B.N.; Raghavendra, S.; Nandi, S.; Dineshkumar, D.; Thayakumar, A.; Parthipan, S.; Ravindra, J.P. Evaluation of maize grain and polyunsaturated fatty acid (PUFA) as energy sources for breeding rams based on hormonal, sperm functional parameters and fertility. Reprod. Fert. Dev. 2012, 24, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Nieto, C.A.R.; Thompson, A.N.; Macleay, C.A.; Briegel, J.R.; Hedger, M.P.; Ferguson, M.B.; Martin, G.B. Relationships among body composition, circulating concentrations of leptin and follistatin, and the onset of puberty and fertility in young female sheep. Anim. Reprod. Sci. 2014, 151, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Dobson, H.; Fergani, C.; Routly, J.E.; Smith, R.F. Effects of stress on reproduction in ewes. Anim. Reprod. Sci. 2012, 130, 135–140. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, J.; Wang, Z.; Yang, H.; Yao, X.; Yang, P.; Ren, C.; Wang, F.; Zhang, Y. Pituitary Transcriptomic Study Reveals the Differential Regulation of lncRNAs and mRNAs Related to Prolificacy in Different FecB Genotyping Sheep. Genes 2019, 10, 157. https://doi.org/10.3390/genes10020157
Zheng J, Wang Z, Yang H, Yao X, Yang P, Ren C, Wang F, Zhang Y. Pituitary Transcriptomic Study Reveals the Differential Regulation of lncRNAs and mRNAs Related to Prolificacy in Different FecB Genotyping Sheep. Genes. 2019; 10(2):157. https://doi.org/10.3390/genes10020157
Chicago/Turabian StyleZheng, Jian, Zhibo Wang, Hua Yang, Xiaolei Yao, Pengcheng Yang, CaiFang Ren, Feng Wang, and YanLi Zhang. 2019. "Pituitary Transcriptomic Study Reveals the Differential Regulation of lncRNAs and mRNAs Related to Prolificacy in Different FecB Genotyping Sheep" Genes 10, no. 2: 157. https://doi.org/10.3390/genes10020157