PheWAS-Based Systems Genetics Methods for Anti-Breast Cancer Drug Discovery


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Sets
2.2. HotNet2 Algorithm
2.3. Gene Dependency Network
2.4. GeneRank Algorithm
2.5. Enrichment Analysis
3. Results and Discussion
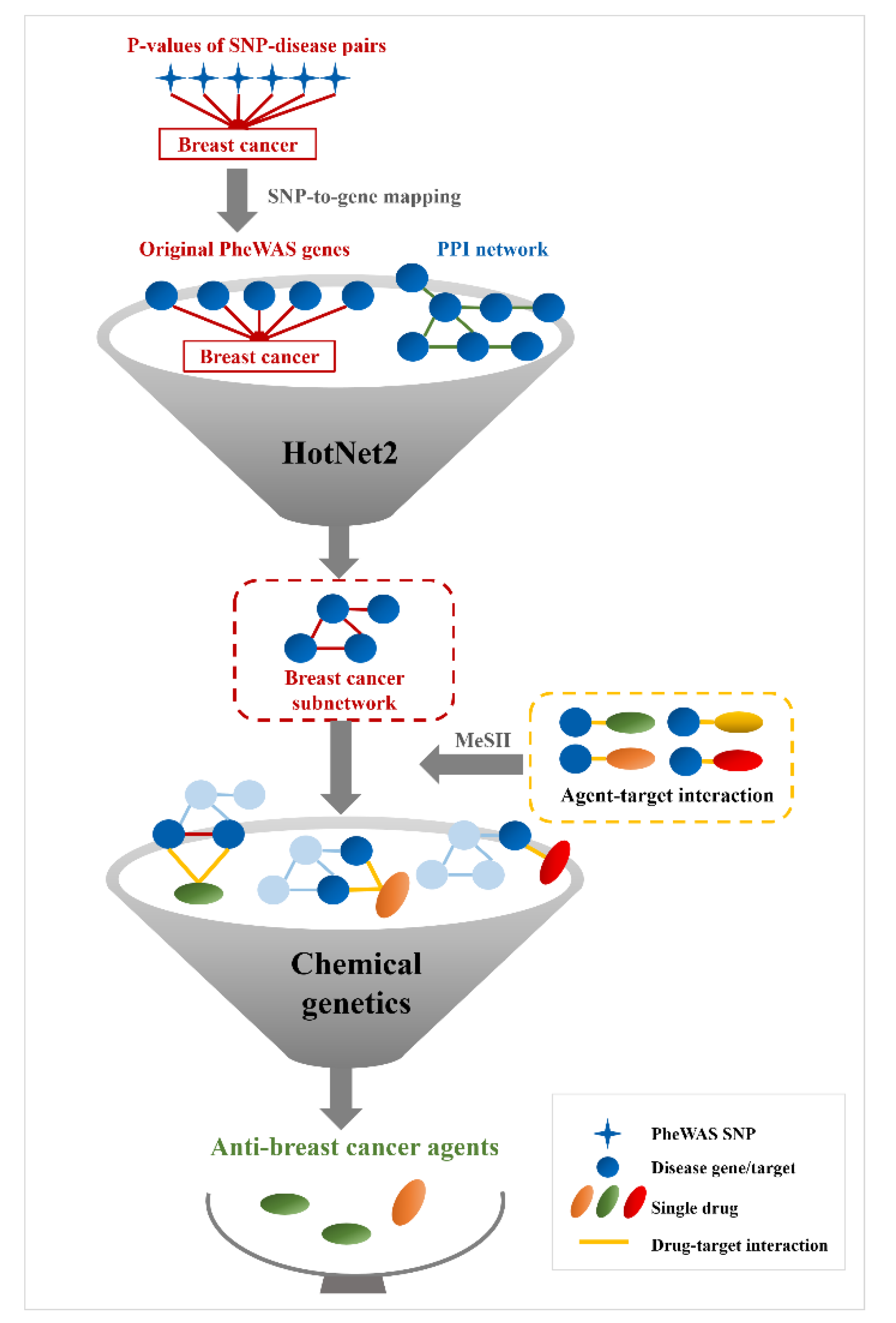
3.1. Identification of Breast Cancer-Associated Genes by HotNet2
3.2. Anti-Breast Cancer Drug Discovery Based on HotNet2-Identified Genes
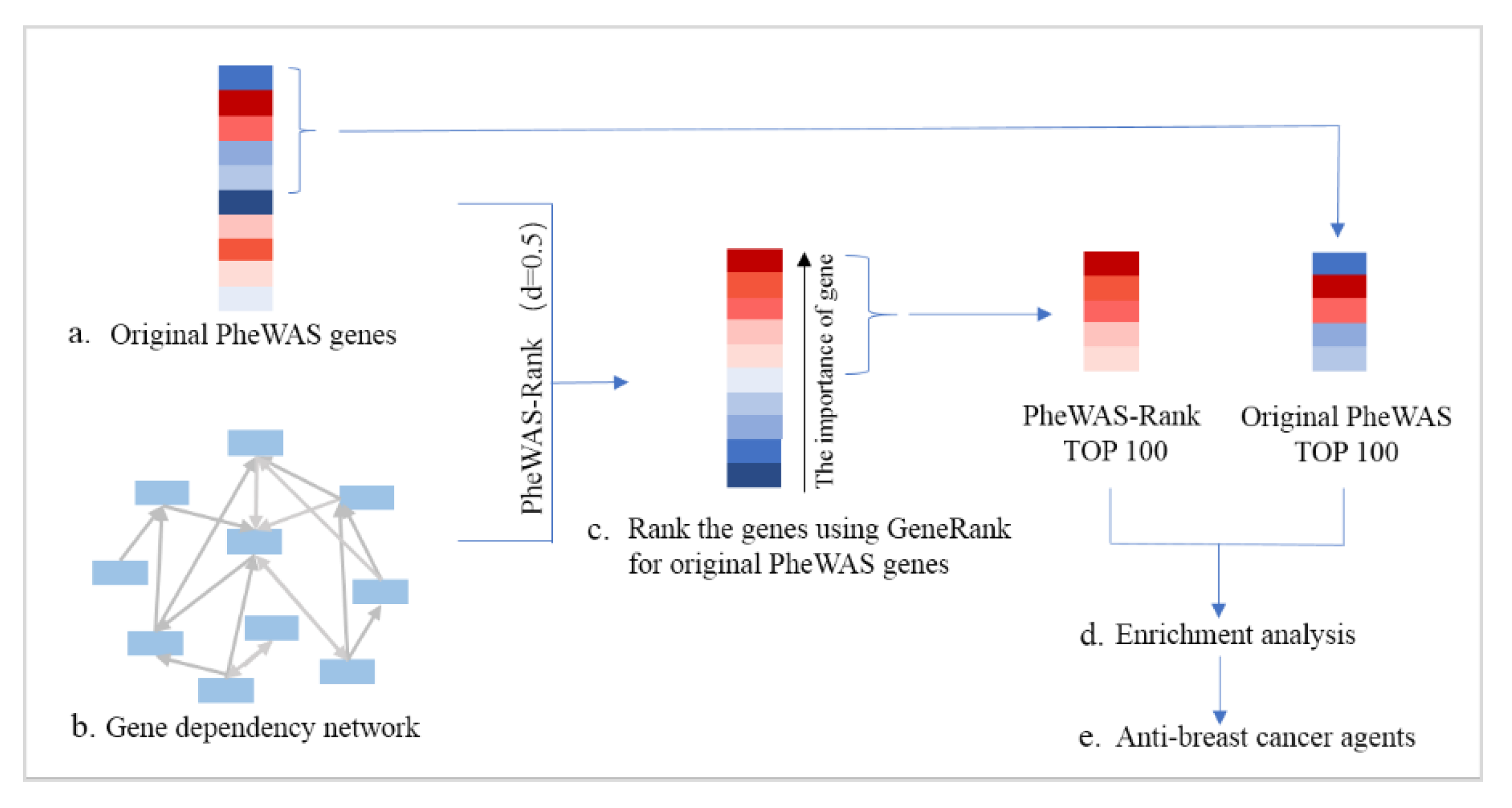
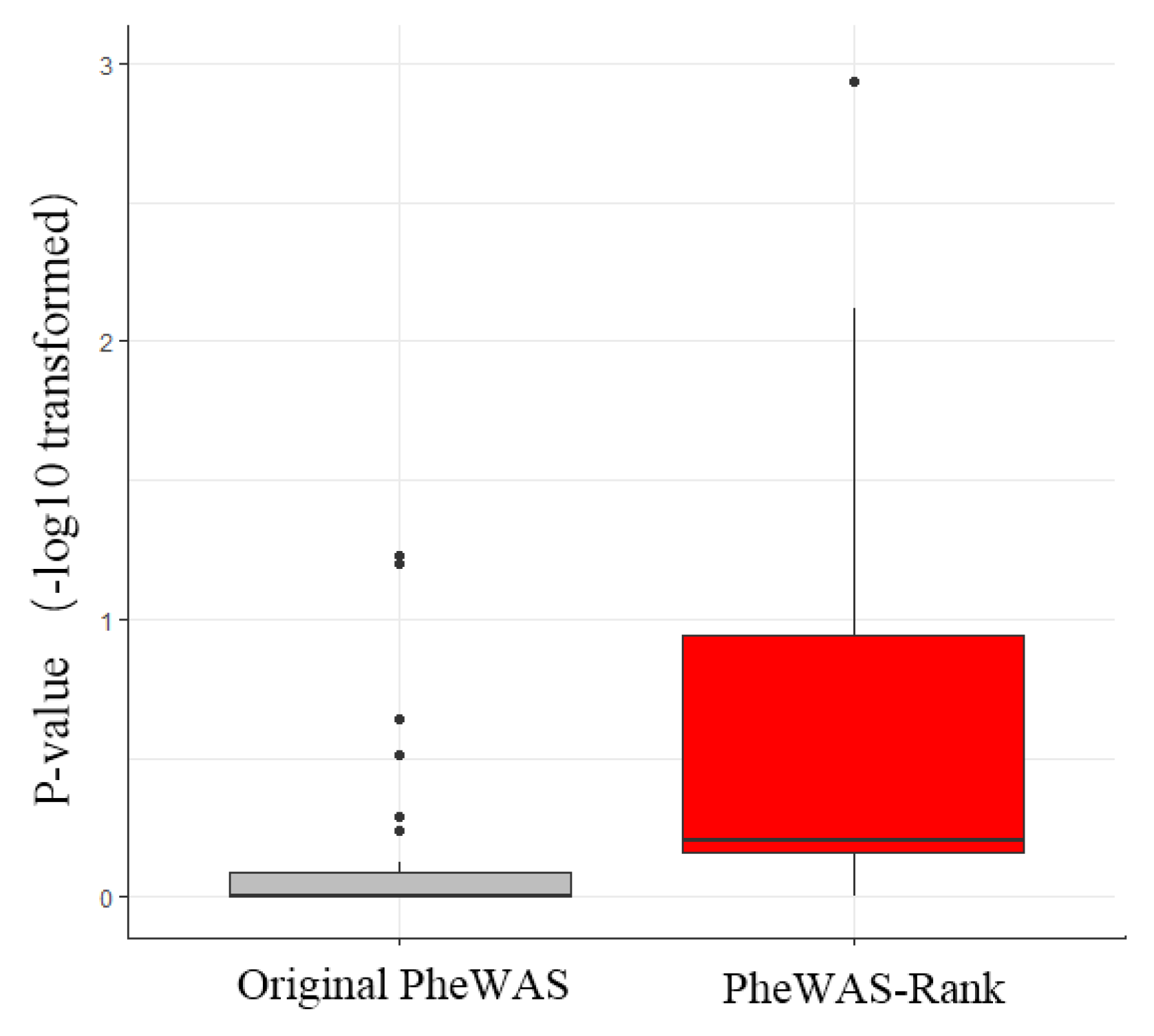
3.3. Identification of Breast Cancer-Associated Genes by PheWAS-Rank
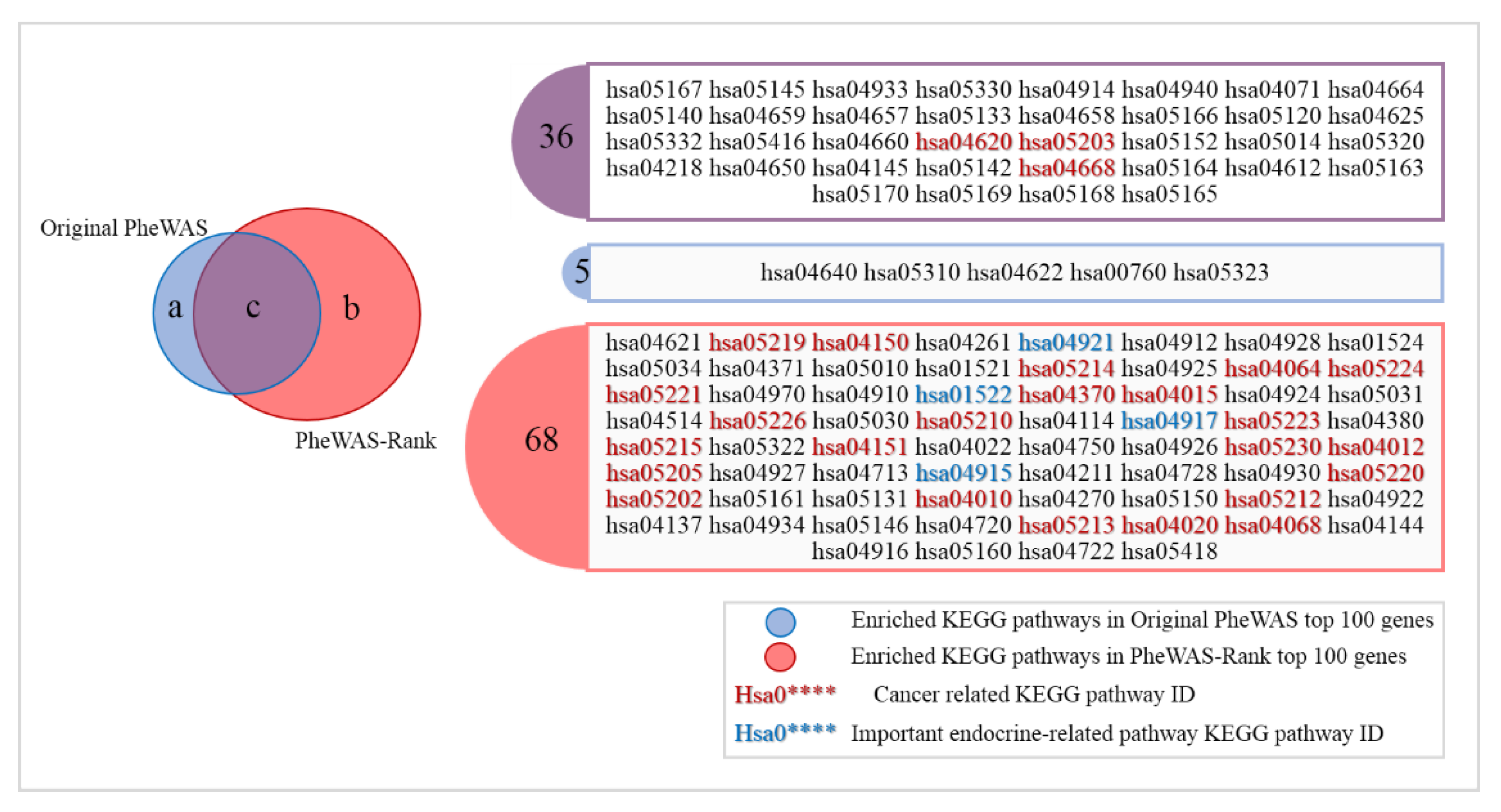
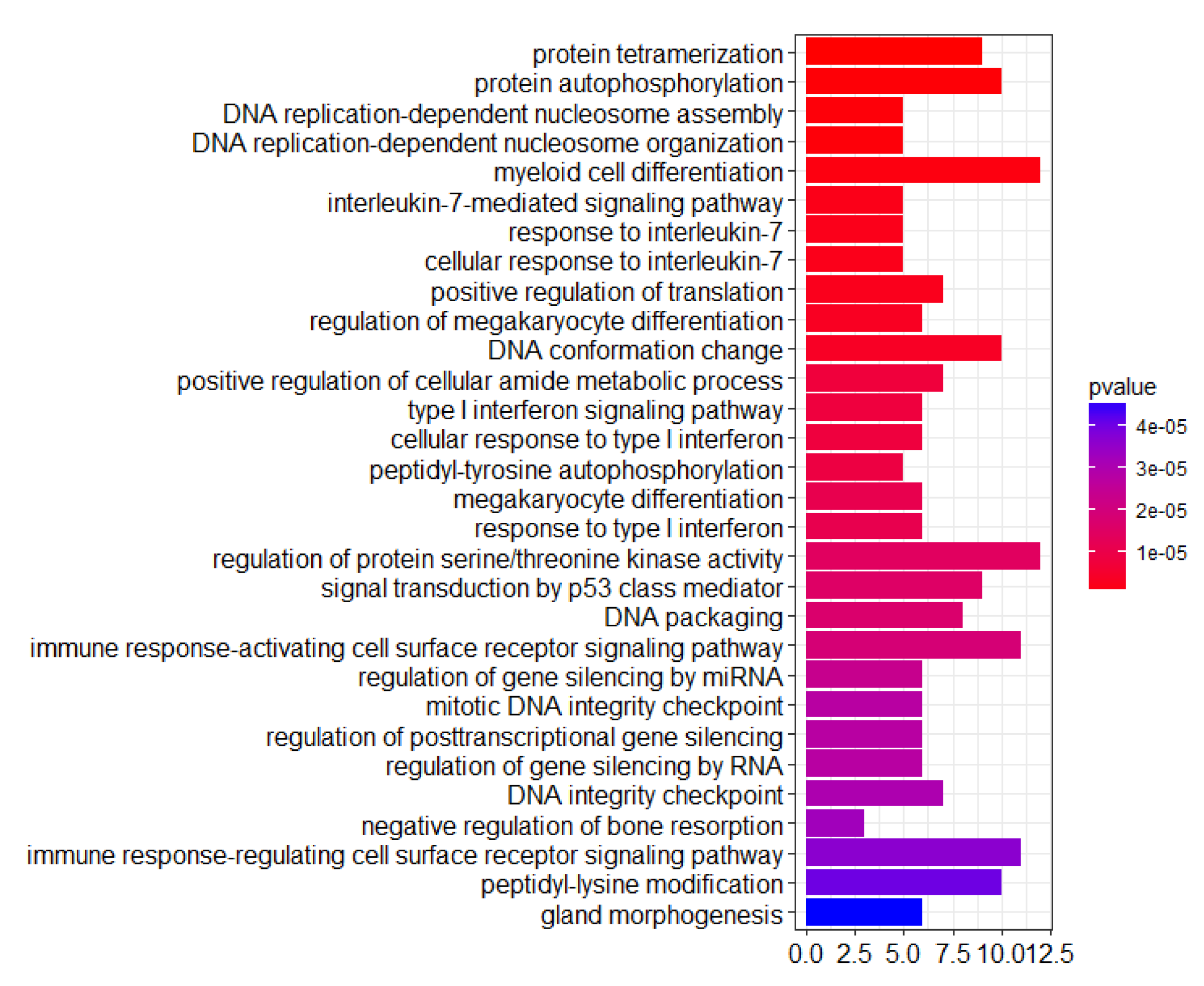
3.4. Anti-Breast Cancer Drug Discovery Based on PheWAS-Rank-Identified Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Boyle, P.; Howell, A. The globalisation of breast cancer. Breast Cancer Res. 2010, 12, S7. [Google Scholar] [CrossRef] [PubMed]
- Scutt, D.; A Lancaster, G.; Manning, J.T. Breast asymmetry and predisposition to breast cancer. Breast Cancer Res. 2006, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Sanseau, P.; Agarwal, P.; Barnes, M.R.; Pastinen, T.; Richards, J.B.; Cardon, L.R.; Mooser, V. Use of genome-wide association studies for drug repositioning. Nat. Biotechnol. 2012, 30, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-Y.; Zhang, H.-Y. Rational drug repositioning by medical genetics. Nat. Biotechnol. 2013, 31, 1080–1082. [Google Scholar] [CrossRef] [PubMed]
- Rastegar-Mojarad, M.; Ye, Z.; Kolesar, J.M.; Hebbring, S.J.; Lin, S.M. Opportunities for drug repositioning from phenome-wide association studies. Nat. Biotechnol. 2015, 33, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Wang, Z.-Y.; Chu, X.-Y.; Zhang, H.-Y. Evolutionary and genetic features of drug targets. Med. Res. Rev. 2018, 38, 1536–1549. [Google Scholar] [CrossRef] [PubMed]
- Denny, J.C.; Bastarache, L.; Ritchie, M.D.; Carroll, R.J.; Zink, R.; Mosley, J.D.; Field, J.R.; Pulley, J.M.; Ramirez, A.H.; Bowton, E.; et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 2013, 31, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Denny, J.C.; Ritchie, M.D.; Basford, M.A.; Pulley, J.M.; Bastarache, L.; Brown-Gentry, K.; Wang, D.; Masys, D.R.; Roden, D.M.; Crawford, D.C.; et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 2010, 26, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Civelek, M.; Lusis, A.J. Systems genetics approaches to understand complex traits. Nat. Rev. Genet. 2013, 15, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.G.; Barnes, K.C.; Bright, T.J.; Wang, S.A. The genetic association database. Nat. Genet. 2004, 36, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2004, 33, D514-7. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2013, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Li, M.J.; Liu, Z.; Wang, P.; Wong, M.P.; Nelson, M.R.; Kocher, J.-P.A.; Yeager, M.; Sham, P.C.; Chanock, S.J.; Xia, Z.; et al. GWASdb v2: an update database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res. 2015, 44, D869–D876. [Google Scholar] [CrossRef] [PubMed]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, A.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2013, 42, D1001–D1006. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, X.; Thijssen, B.; Das, J.; Lipkin, S.M.; Yu, H. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat. Biotechnol. 2012, 30, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Aronson, A.R. Effective mapping of biomedical text to the UMLS Metathesaurus: The MetaMap program. Proc. AMIA Symp. 2001, 17–21. [Google Scholar]
- Wagner, A.H.; Coffman, A.C.; Ainscough, B.J.; Spies, N.C.; Skidmore, Z.L.; Campbell, K.M.; Krysiak, K.; Pan, D.; McMichael, J.F.; Eldred, J.M.; et al. DGIdb 2.0: mining clinically relevant drug–gene interactions. Nucleic Acids Res. 2015, 44, D1036–D1044. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhang, C.; Zhu, F.; Xu, F.; Chen, S.Y.; Zhang, P.; Li, Y.H.; Yang, S.Y.; Wei, Y.Q.; Tao, L.; et al. Therapeutic target database update 2014: a resource for targeted therapeutics. Nucleic Acids Res. 2013, 42, D1118–D1123. [Google Scholar] [CrossRef] [PubMed]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2013, 42, D1091–D1097. [Google Scholar] [CrossRef] [PubMed]
- Leiserson, M.D.M.; Vandin, F.; Wu, H.-T.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; McLellan, M.; et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2014, 47, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, J. Inferring Gene Dependency Network Specific to Phenotypic Alteration Based on Gene Expression Data and Clinical Information of Breast Cancer. PLoS ONE 2014, 9, e92023. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.L.; Breitling, R.; Higham, D.J.; Gilbert, D.R. GeneRank: using search engine technology for the analysis of microarray experiments. BMC Bioinform. 2005, 6, 233. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Y.; Chen, L.-L.; Zhou, X.-H. Identifying prognostic signature in ovarian cancer using DirGenerank. Oncotarget 2017, 8, 46398–46413. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.A.; Carnevale, R.P.; Proietti, C.J.; Rosemblit, C.; Beguelin, W.; Salatino, M.; Charreau, E.H.; Frahm, I.; Sapia, S.; Brouckaert, P.; et al. TNF α acting on TNFR1 promotes breast cancer growth via p42/p44 MAPK, JNK, Akt and NF-kappa B-dependent pathways. Exp. Cell Res. 2008, 314, 509–529. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, J.; Luo, J.; Lai, F.; Wang, Z.; Tong, H.; Lu, D.; Bu, H.; Zhang, R.; Lin, S. Antiangiogenic effects of oxymatrine on pancreatic cancer by inhibition of the NF-κB-mediated VEGF signaling pathway. Oncol. Rep. 2013, 30, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-C.; Hsu, H.-M.; Chen, S.-T.; Hsu, G.-C.; Huang, C.-S.; Hou, M.-F.; Fu, Y.-P.; Cheng, T.-C.; Wu, P.-E.; Shen, C.-Y.; et al. Breast cancer risk associated with genotypic polymorphism of the genes involved in the estrogen-receptor-signaling pathway: a multigenic study on cancer susceptibility. J. Biomed. Sci. 2006, 13, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Llovera, M.; Pichard, C.; Bernichtein, S.; Jeay, S.; Touraine, P.; Kelly, P.A.; Goffin, V. Human prolactin (hPRL) antagonists inhibit hPRL-activated signaling pathways involved in breast cancer cell proliferation. Oncogene 2000, 19, 4695–4705. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.M.; Heydari, Z.; Rahimi, M.; Bazgir, B.; Shirvani, H.; Alipour, S.; Heidarian, Y.; Khalighfard, S.; Isanejad, A. Oxytocin mediates the beneficial effects of the exercise training on breast cancer. Exp. Physiol. 2017, 103, 222–235. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, M.; Quan, Y.; Zhou, X.-H.; Zhang, H.-Y. PheWAS-Based Systems Genetics Methods for Anti-Breast Cancer Drug Discovery. Genes 2019, 10, 154. https://doi.org/10.3390/genes10020154
Gao M, Quan Y, Zhou X-H, Zhang H-Y. PheWAS-Based Systems Genetics Methods for Anti-Breast Cancer Drug Discovery. Genes. 2019; 10(2):154. https://doi.org/10.3390/genes10020154
Chicago/Turabian StyleGao, Min, Yuan Quan, Xiong-Hui Zhou, and Hong-Yu Zhang. 2019. "PheWAS-Based Systems Genetics Methods for Anti-Breast Cancer Drug Discovery" Genes 10, no. 2: 154. https://doi.org/10.3390/genes10020154