Dissecting the Genome-Wide Evolution and Function of R2R3-MYB Transcription Factor Family in Rosa chinensis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Growth and Sample Collection
2.2. Plant Genome Analysis and Pairwise Whole-Genome Comparisons
2.3. Identification of R2R3-MYB Gene Models
2.4. Calculation of Syntenic Blocks and Construction of a Synteny Network for R2R3-MYBs
2.5. Network Clustering
2.6. Chromosomal Localization and Phylogenetic Analysis
2.7. Calculation of Ka/Ks Ratios of Gene Pairs
2.8. Transcriptome Sequencing, Functional Annotation, and Analysis of Differentially Expressed Genes
2.9. Validation of Gene Expression by a qRT-PCR Assay
2.10. Transient Overexpression of RcMYB114a in Tobacco
3. Results
3.1. Identification of R2R3-MYB Genes in 35 Genomes
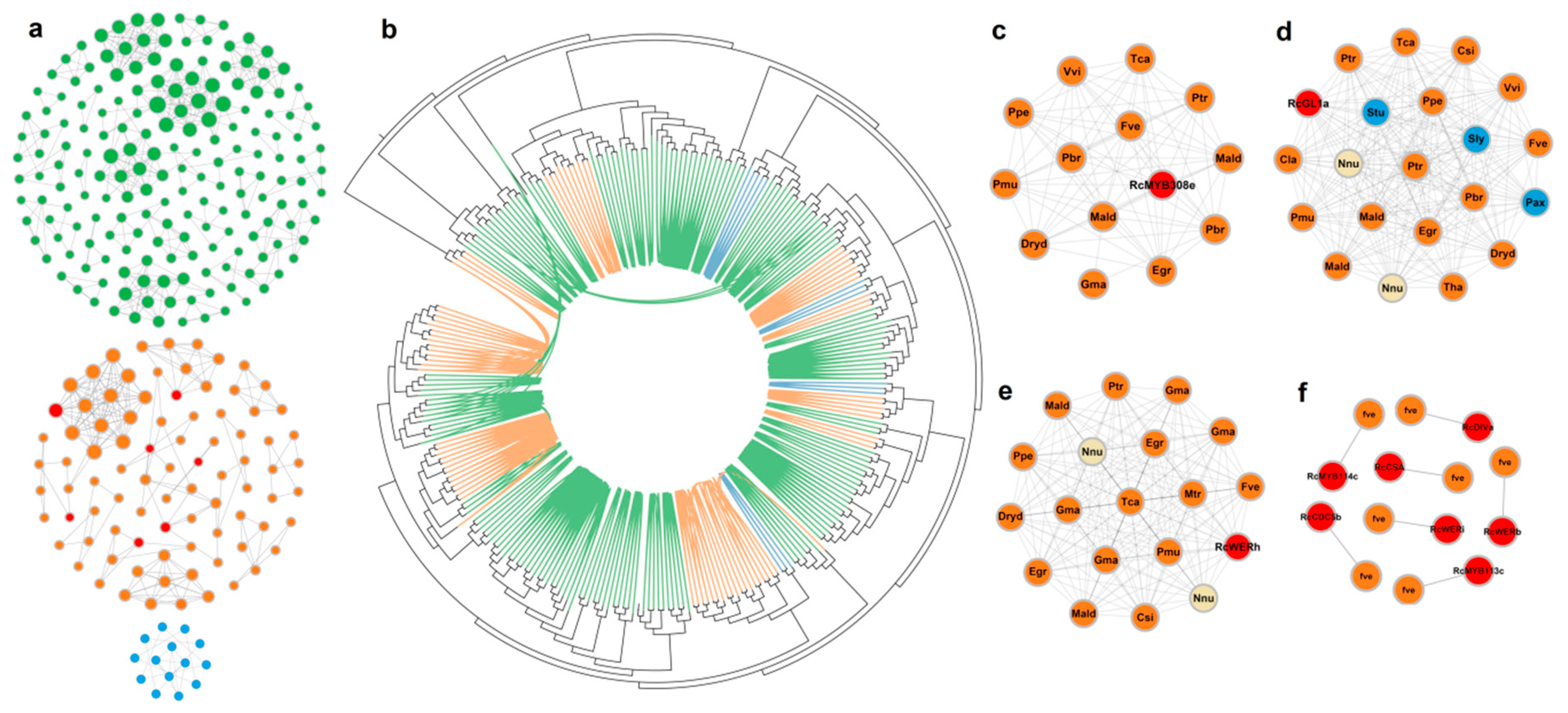
3.2. Some Lineage-Specific Clusters of R2R3-MYBs
3.3. Physical Distribution of R2R3-MYB Genes on R. chinensis Chromosomes
3.4. Predicted Functions and Analysis of the Spatiotemporal Expression of R2R3-MYB Genes in R. chinensis
3.5. RcMYB114a May Affect the Formation of the Flower Color and Fragrance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Riechmann, J.L.; Heard, J.; Martin, G.; Reuber, L.; Jiang, C.-Z.; Keddie, J.; Adam, L.; Pineda, O.; Ratcliffe, O.; Samaha, R. Arabidopsis transcription factors: Genome-wide comparative analysis among eukaryotes. Science 2000, 290, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Mmadi, M.; Dossa, K.; Wang, L.; Zhou, R.; Wang, Y.; Cisse, N.; Sy, M.; Zhang, X. Functional characterization of the versatile MYB gene family uncovered their important roles in plant development and responses to drought and waterlogging in sesame. Genes 2017, 8, 362. [Google Scholar] [CrossRef] [PubMed]
- Rogers, L.A.; Campbell, M.M. The genetic control of lignin deposition during plant growth and development. New Phytol. 2004, 164, 17–30. [Google Scholar] [CrossRef]
- Du, H.; Zhang, L.; Liu, L.; Tang, X.F.; Yang, W.J.; Wu, Y.M.; Huang, Y.B.; Tang, Y.X. Biochemical and molecular characterization of plant MYB transcription factor family. Biochemistry (Moscow) 2009, 74, 1–11. [Google Scholar] [CrossRef]
- Feller, A.; Machemer, K.; Braun, E.L.; Grotewold, E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011, 66, 94–116. [Google Scholar] [CrossRef]
- Jin, H.; Martin, C. Multifunctionality and diversity within the plant MYB-gene family. Plant Mol. Biol. 1999, 41, 577–585. [Google Scholar] [CrossRef]
- Urao, T.; Yamaguchi-Shinozaki, K.; Urao, S.; Shinozaki, K. An Arabidopsis myb homolog is induced by dehydration stress and its gene product binds to the conserved MYB recognition sequence. Plant Cell 1993, 5, 1529–1539. [Google Scholar]
- Abe, H.; Yamaguchi-Shinozaki, K.; Urao, T.; Iwasaki, T.; Hosokawa, D.; Shinozaki, K. Role of Arabidopsis MYC and MYB homologs in drought-and abscisic acid-regulated gene expression. Plant Cell 1997, 9, 1859–1868. [Google Scholar]
- Gocal, G.F.; Sheldon, C.C.; Gubler, F.; Moritz, T.; Bagnall, D.J.; MacMillan, C.P.; Li, S.F.; Parish, R.W.; Dennis, E.S.; Weigel, D. GAMYB-like genes, flowering, and gibberellin signaling in Arabidopsis. Plant Physiol. 2001, 127, 1682–1693. [Google Scholar] [CrossRef]
- Jin, H.; Cominelli, E.; Bailey, P.; Parr, A.; Mehrtens, F.; Jones, J.; Tonelli, C.; Weisshaar, B.; Martin, C. Transcriptional repression by AtMYB4 controls production of UV-protecting sunscreens in Arabidopsis. EMBO J. 2000, 19, 6150–6161. [Google Scholar] [CrossRef] [PubMed]
- Urao, T.; Noji, M.A.; Yamaguchi-Shinozaki, K.; Shinozaki, K. A transcriptional activation domain of ATMYB2, a drought-inducible Arabidopsis Myb-related protein. Plant J. 1996, 10, 1145–1148. [Google Scholar] [CrossRef]
- Liu, J.; Osbourn, A.; Ma, P. MYB transcription factors as regulators of phenylpropanoid metabolism in plants. Mol. Plant 2015, 8, 689–708. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, J.; Ghosal, D.; Wienand, U.; Peterson, P.; Saedler, H. The regulatory c1 locus of Zea mays encodes a protein with homology to myb proto-oncogene products and with structural similarities to transcriptional activators. EMBO J. 1987, 6, 3553–3558. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, O.; Nahal, H.; Foong, J.; Provart, N.J.; Campbell, M.M. Expansion and diversification of the Populus R2R3-MYB family of transcription factors. Plant Physiol. 2009, 149, 981–993. [Google Scholar] [CrossRef]
- Yanhui, C.; Xiaoyuan, Y.; Kun, H.; Meihua, L.; Jigang, L.; Zhaofeng, G.; Zhiqiang, L.; Yunfei, Z.; Xiaoxiao, W.; Xiaoming, Q. The MYB transcription factor superfamily of Arabidopsis: Expression analysis and phylogenetic comparison with the rice MYB family. Plant Mol. Biol. 2006, 60, 107–124. [Google Scholar] [CrossRef]
- Du, H.; Feng, B.-R.; Yang, S.-S.; Huang, Y.-B.; Tang, Y.-X. The R2R3-MYB transcription factor gene family in maize. PLoS ONE 2012, 7, e37463. [Google Scholar] [CrossRef]
- Du, H.; Yang, S.-S.; Liang, Z.; Feng, B.-R.; Liu, L.; Huang, Y.-B.; Tang, Y.-X. Genome-wide analysis of the MYB transcription factor superfamily in soybean. BMC Plant Biol. 2012, 12, 106. [Google Scholar] [CrossRef]
- Cao, Z.-H.; Zhang, S.-Z.; Wang, R.-K.; Zhang, R.-F.; Hao, Y.-J. Genome wide analysis of the apple MYB transcription factor family allows the identification of MdoMYB121 gene confering abiotic stress tolerance in plants. PLoS ONE 2013, 8, e69955. [Google Scholar] [CrossRef]
- Soler, M.; Camargo, E.L.O.; Carocha, V.; Cassan-Wang, H.; San Clemente, H.; Savelli, B.; Hefer, C.A.; Paiva, J.A.P.; Myburg, A.A.; Grima-Pettenati, J. The Eucalyptus grandis R2R3-MYB transcription factor family: Evidence for woody growth-related evolution and function. New Phytol. 2015, 206, 1364–1377. [Google Scholar] [CrossRef]
- Jiang, C.; Gu, X.; Peterson, T. Identification of conserved gene structures and carboxy-terminal motifs in the Myb gene family of Arabidopsis and Oryza sativa L. ssp. indica. Genome Biol. 2004, 5, R46. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, X.; Xu, Y.; Deng, X.; Xu, Q. Genome-wide analysis of the R2R3-MYB transcription factor gene family in sweet orange (Citrus sinensis). Mol. Biol. Rep. 2014, 41, 6769–6785. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Tang, H.; Wang, X.; Paterson, A.H. PGDD: A database of gene and genome duplication in plants. Nucleic Acids Res. 2012, 41, D1152–D1158. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Li, J.; Tang, H.; Paterson, A.H. Integrated syntenic and phylogenomic analyses reveal an ancient genome duplication in monocots. Plant Cell 2014, 26, 2792–2802. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C. Modern Roses XI: The World Encyclopedia of Roses; American Rose Society: Shreveport, LA, USA, 2000; p. 638. [Google Scholar]
- Raymond, O.; Gouzy, J.; Just, J.; Badouin, H.; Verdenaud, M.; Lemainque, A.; Vergne, P.; Moja, S.; Choisne, N.; Pont, C. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 2018, 50, 772. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef]
- Falcone Ferreyra, M.L.; Rius, S.; Casati, P. Flavonoids: Biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 2012, 3, 222. [Google Scholar] [CrossRef]
- Zvi, M.M.B.; Shklarman, E.; Masci, T.; Kalev, H.; Debener, T.; Shafir, S.; Ovadis, M.; Vainstein, A. PAP1 transcription factor enhances production of phenylpropanoid and terpenoid scent compounds in rose flowers. New Phytol. 2012, 195, 335–345. [Google Scholar] [CrossRef]
- Bateman, A.; Coin, L.; Durbin, R.; Finn, R.D.; Hollich, V.; Griffiths-Jones, S.; Khanna, A.; Marshall, M.; Moxon, S.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res. 2004, 32, D138–D141. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Zhao, T.; Schranz, M.E. Network approaches for plant phylogenomic synteny analysis. Curr. Opin. Plant Biol. 2017, 36, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. Icwsm 2009, 8, 361–362. [Google Scholar]
- Palla, G.; Derényi, I.; Farkas, I.; Vicsek, T. Uncovering the overlapping community structure of complex networks in nature and society. Nature 2005, 435, 814. [Google Scholar] [CrossRef]
- Fortunato, S. Community detection in graphs. Phys. Rep. 2010, 486, 75–174. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Voorrips, R. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq- a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression of RNA-Seq data at the gene level–the DESeq package. Heidelb. Ger. Eur. Mol. Biol. Lab. (EMBL) 2012. Available online: www.bioconductor.org/packages/devel/bioc/vignettes/DESeq/inst/doc/DESeq.pdf (accessed on 17 October 2019).
- Han, Y.; Wan, H.; Cheng, T.; Wang, J.; Yang, W.; Pan, H.; Zhang, Q. Comparative RNA-seq analysis of transcriptome dynamics during petal development in Rosa chinensis. Sci. Rep. 2017, 7, 43382. [Google Scholar] [CrossRef]
- Zhao, T.; Holmer, R.; de Bruijn, S.; Angenent, G.C.; van den Burg, H.A.; Schranz, M.E. Phylogenomic synteny network analysis of MADS-box transcription factor genes reveals lineage-specific transpositions, ancient tandem duplications, and deep positional conservation. Plant Cell 2017, 29, 1278–1292. [Google Scholar] [CrossRef]
- Scalliet, G.; Journot, N.; Jullien, F.; Baudino, S.; Magnard, J.-L.; Channelière, S.; Vergne, P.; Dumas, C.; Bendahmane, M.; Cock, J.M. Biosynthesis of the major scent components 3, 5-dimethoxytoluene and 1, 3, 5-trimethoxybenzene by novel rose O-methyltransferases. FEBS Lett. 2002, 523, 113–118. [Google Scholar] [CrossRef]
- Lavid, N.; Wang, J.; Shalit, M.; Guterman, I.; Bar, E.; Beuerle, T.; Menda, N.; Shafir, S.; Zamir, D.; Adam, Z. O-methyltransferases involved in the biosynthesis of volatile phenolic derivatives in rose petals. Plant Physiol. 2002, 129, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Conant, G.C.; Wolfe, K.H. Turning a hobby into a job: How duplicated genes find new functions. Nat. Rev. Genet. 2008, 9, 938. [Google Scholar] [CrossRef] [PubMed]
- Flagel, L.E.; Wendel, J.F. Gene duplication and evolutionary novelty in plants. New Phytol. 2009, 183, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shi, Z.; Maximova, S.N.; Payne, M.J.; Guiltinan, M.J. Tc-MYBPA is an Arabidopsis TT2-like transcription factor and functions in the regulation of proanthocyanidin synthesis in Theobroma cacao. BMC Plant Biol. 2015, 15, 160. [Google Scholar] [CrossRef] [PubMed]
- Quattrocchio, F.; Wing, J.F.; Leppen, H.T.; Mol, J.N.; Koes, R.E. Regulatory genes controlling anthocyanin pigmentation are functionally conserved among plant species and have distinct sets of target genes. Plant Cell 1993, 5, 1497–1512. [Google Scholar] [CrossRef] [PubMed]
- Albert, N.W.; Lewis, D.H.; Zhang, H.; Schwinn, K.E.; Jameson, P.E.; Davies, K.M. Members of an R2R3-MYB transcription factor family in Petunia are developmentally and environmentally regulated to control complex floral and vegetative pigmentation patterning. Plant J. 2011, 65, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Ming, M.; Allan, A.C.; Gu, C.; Li, L.; Wu, X.; Wang, R.; Chang, Y.; Qi, K.; Zhang, S. Map-based cloning of the pear gene MYB 114 identifies an interaction with other transcription factors to coordinately regulate fruit anthocyanin biosynthesis. Plant J. 2017, 92, 437–451. [Google Scholar] [CrossRef]
- Wei, X.; Liu, K.; Zhang, Y.; Feng, Q.; Wang, L.; Zhao, Y.; Li, D.; Zhao, Q.; Zhu, X.; Zhu, X. Genetic discovery for oil production and quality in sesame. Nat. Commun. 2015, 6, 8609. [Google Scholar] [CrossRef]
- Bhatia, C.; Pandey, A.; Gaddam, S.R.; Hoecker, U.; Trivedi, P.K. Low temperature-enhanced flavonol synthesis requires light-associated regulatory components in Arabidopsis thaliana. Plant Cell Physiol. 2018, 59, 2099–2112. [Google Scholar] [CrossRef]
- Scalliet, G.; Piola, F.; Douady, C.J.; Réty, S.; Raymond, O.; Baudino, S.; Bordji, K.; Bendahmane, M.; Dumas, C.; Cock, J.M. Scent evolution in Chinese roses. Proc. Natl. Acad. Sci. USA 2008, 105, 5927–5932. [Google Scholar] [CrossRef] [Green Version]
- Dewey, C.N. Positional orthology: Putting genomic evolutionary relationships into context. Brief. Bioinform. 2011, 12, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Altenhoff, A.M.; Boeckmann, B.; Capella-Gutierrez, S.; Dalquen, D.A.; DeLuca, T.; Forslund, K.; Huerta-Cepas, J.; Linard, B.; Pereira, C.; Pryszcz, L.P. Standardized benchmarking in the quest for orthologs. Nat. Methods 2016, 13, 425–430. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Y.; Yu, J.; Zhao, T.; Cheng, T.; Wang, J.; Yang, W.; Pan, H.; Zhang, Q. Dissecting the Genome-Wide Evolution and Function of R2R3-MYB Transcription Factor Family in Rosa chinensis. Genes 2019, 10, 823. https://doi.org/10.3390/genes10100823
Han Y, Yu J, Zhao T, Cheng T, Wang J, Yang W, Pan H, Zhang Q. Dissecting the Genome-Wide Evolution and Function of R2R3-MYB Transcription Factor Family in Rosa chinensis. Genes. 2019; 10(10):823. https://doi.org/10.3390/genes10100823
Chicago/Turabian StyleHan, Yu, Jiayao Yu, Tao Zhao, Tangren Cheng, Jia Wang, Weiru Yang, Huitang Pan, and Qixiang Zhang. 2019. "Dissecting the Genome-Wide Evolution and Function of R2R3-MYB Transcription Factor Family in Rosa chinensis" Genes 10, no. 10: 823. https://doi.org/10.3390/genes10100823