Therapeutic Benefit of the Association of Lodenafil with Mesenchymal Stem Cells on Hypoxia-induced Pulmonary Hypertension in Rats

 , ,
, ,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs and Reagents
2.2. Isolation and Immunophenotyping of Umbilical Cord hSMCs (hMSCs)
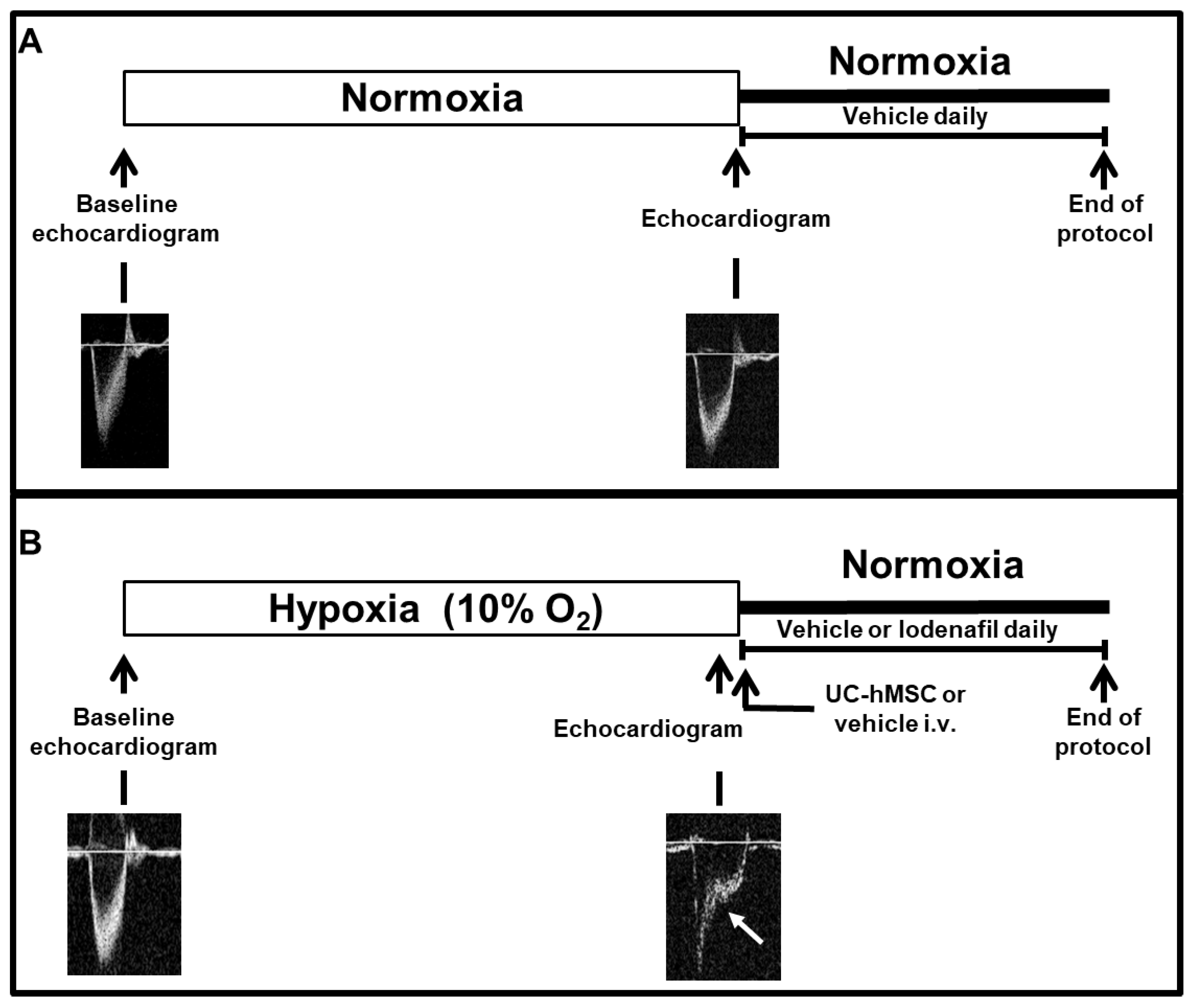
2.3. Experimental Design
- SuHx + vehicle (DMSO p.o.);
- SuHx + lodenafil (10 mg/kg/day p.o.);
- SuHx + hMSCs (5.105 cells i.v.);
- SuHx + lodenafil (10 mg/kg/day p.o.) + hMSCs (5.105 cells i.v.).
2.4. Echocardiography
2.5. RV Catheterization
2.6. Tissue Harvesting
2.7. Vascular Reactivity of Pulmonary Artery
2.8. Membrane Preparation and Western Blot Analysis
2.9. Histological and Immunohistochemistry Analysis of Pulmonary Arterioles and RV
2.10. Statistical Analysis
3. Results
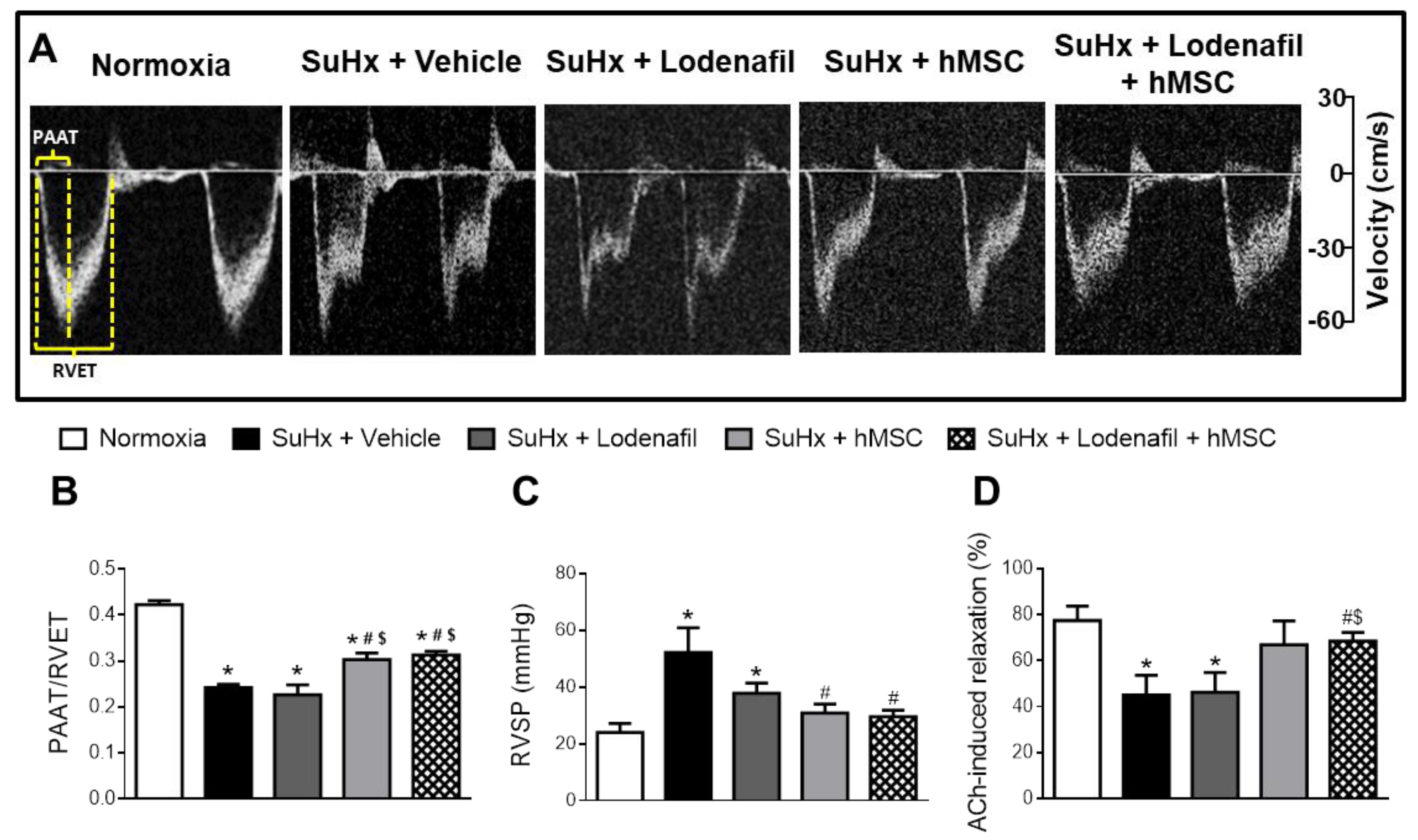
3.1. Lodenafil + hMSCs Reduces Vascular Dysfunction on SuHx-Induced PAH
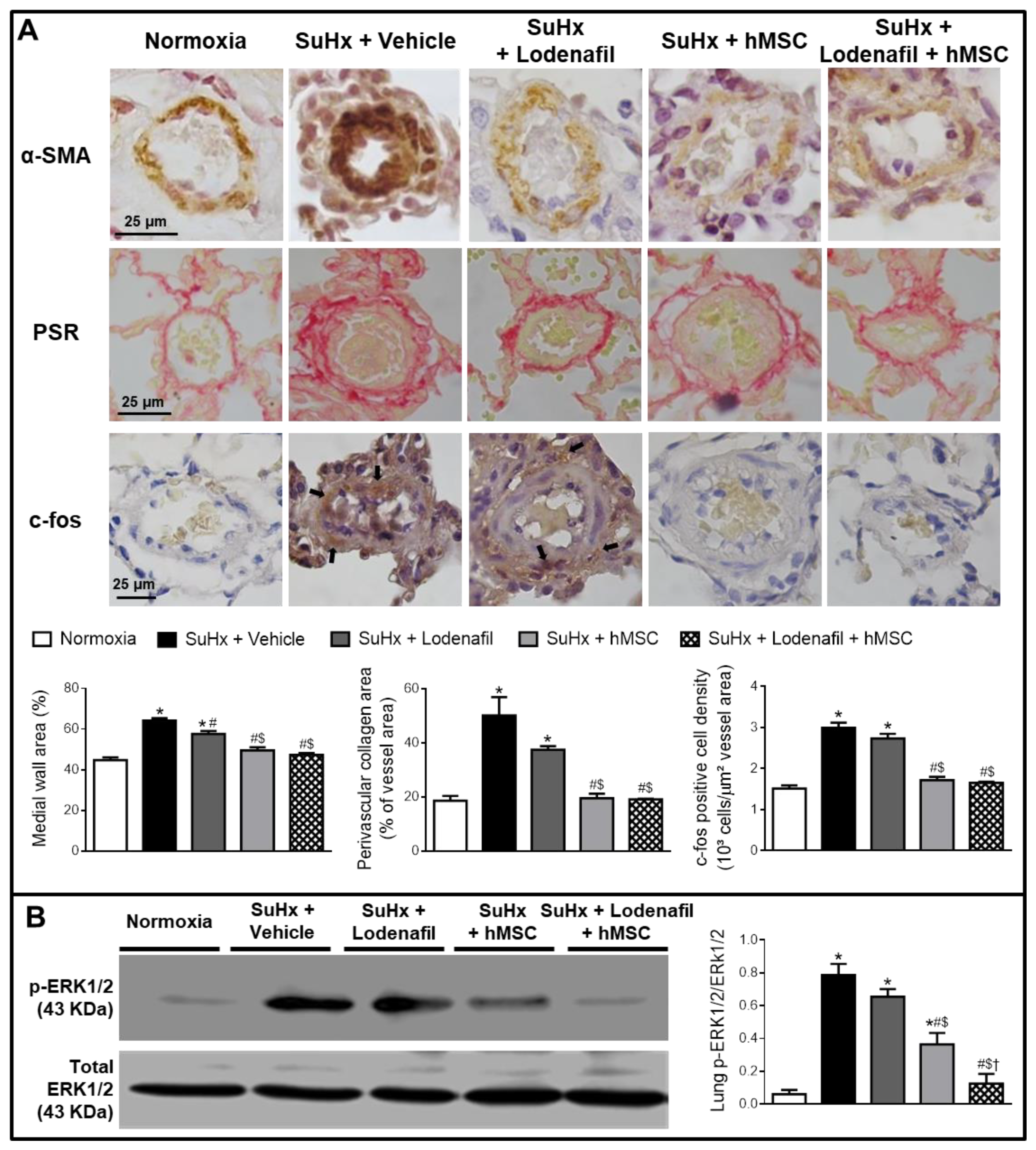
3.2. Lodenafil + hMSCs Reduces Changes in Lungs from SuHx-PAH Rats
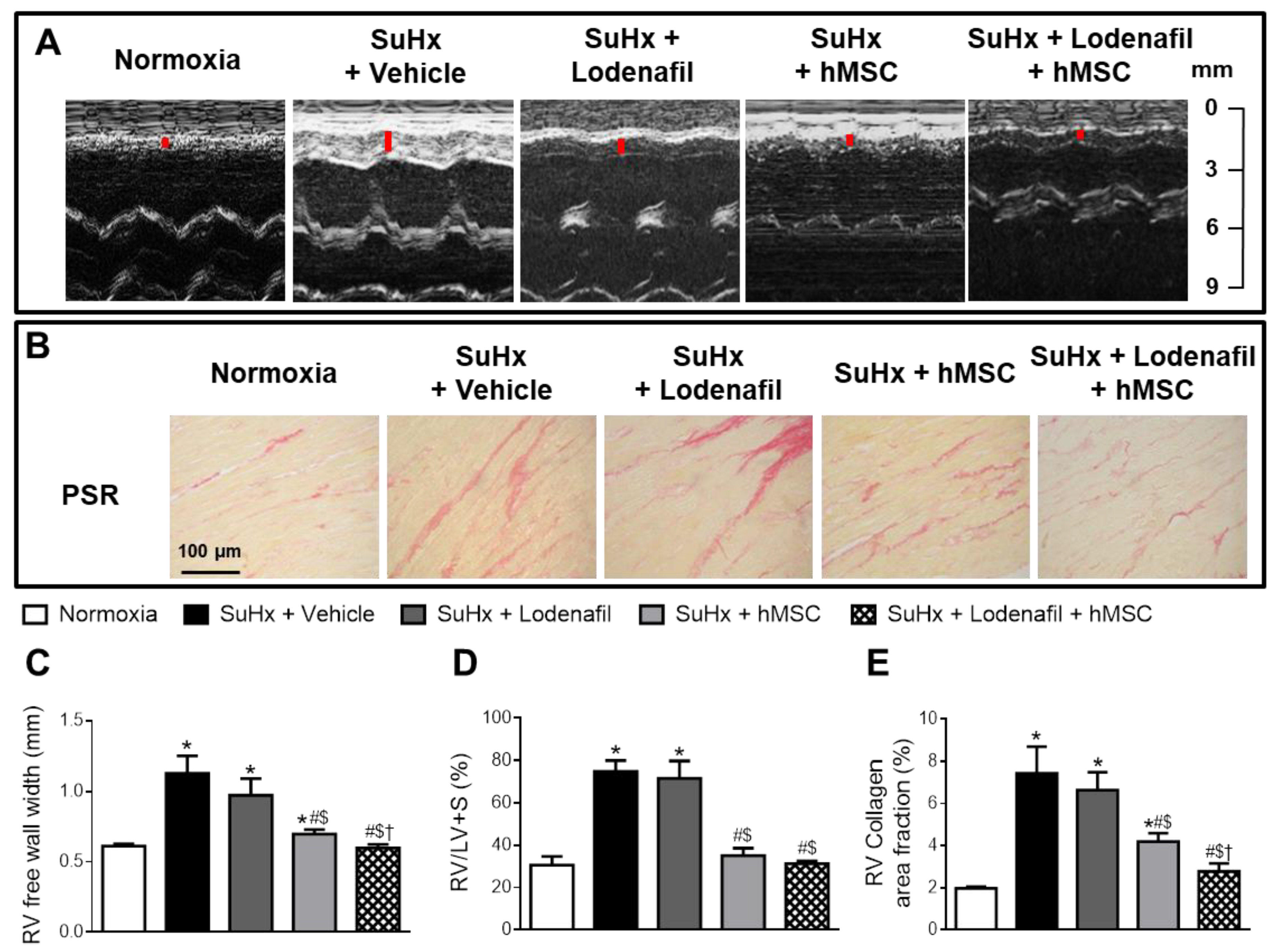
3.3. Lodenafil + hMSCs Therapy Reverses Structural Changes in RV from SuHx-PAH
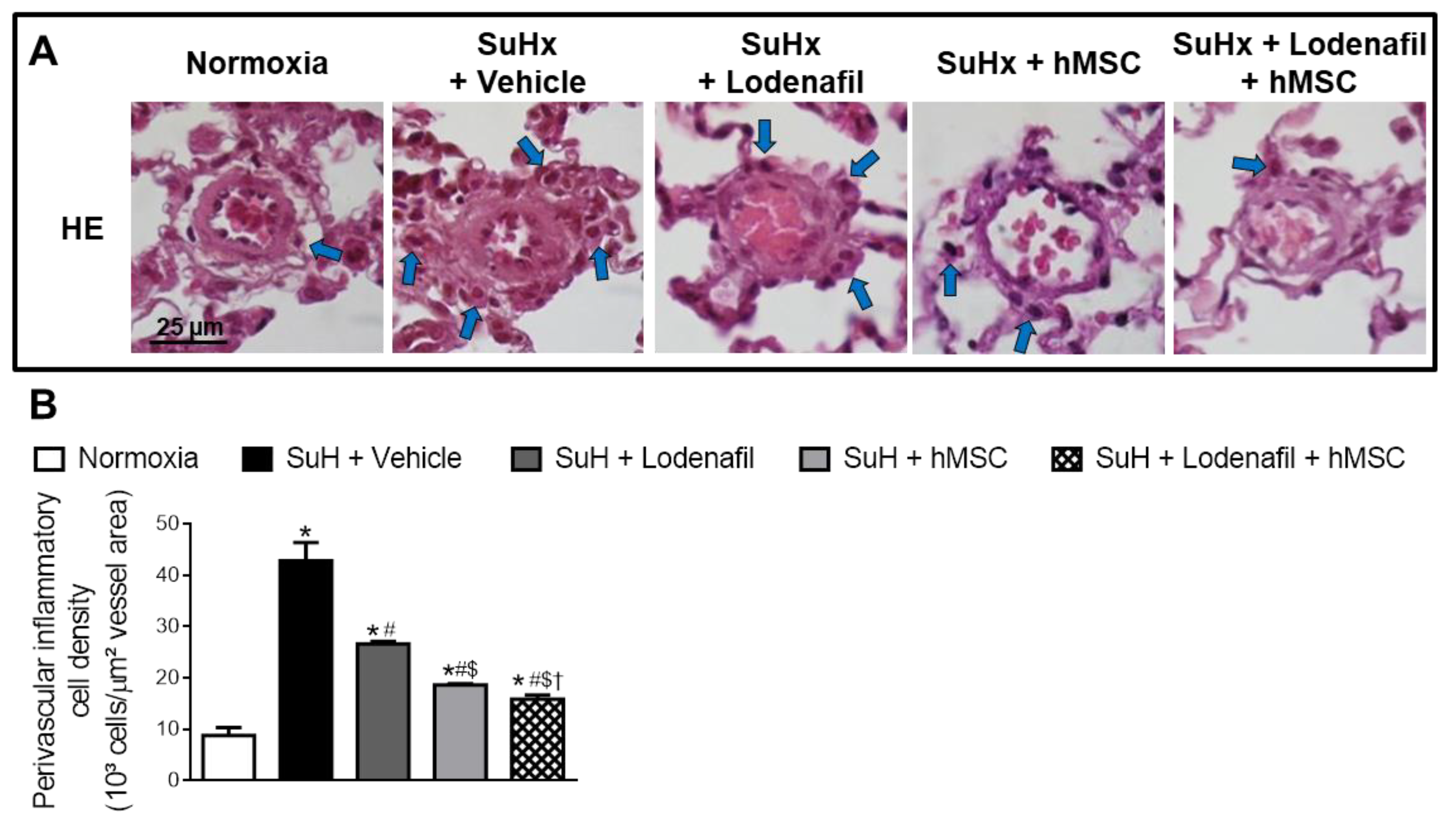
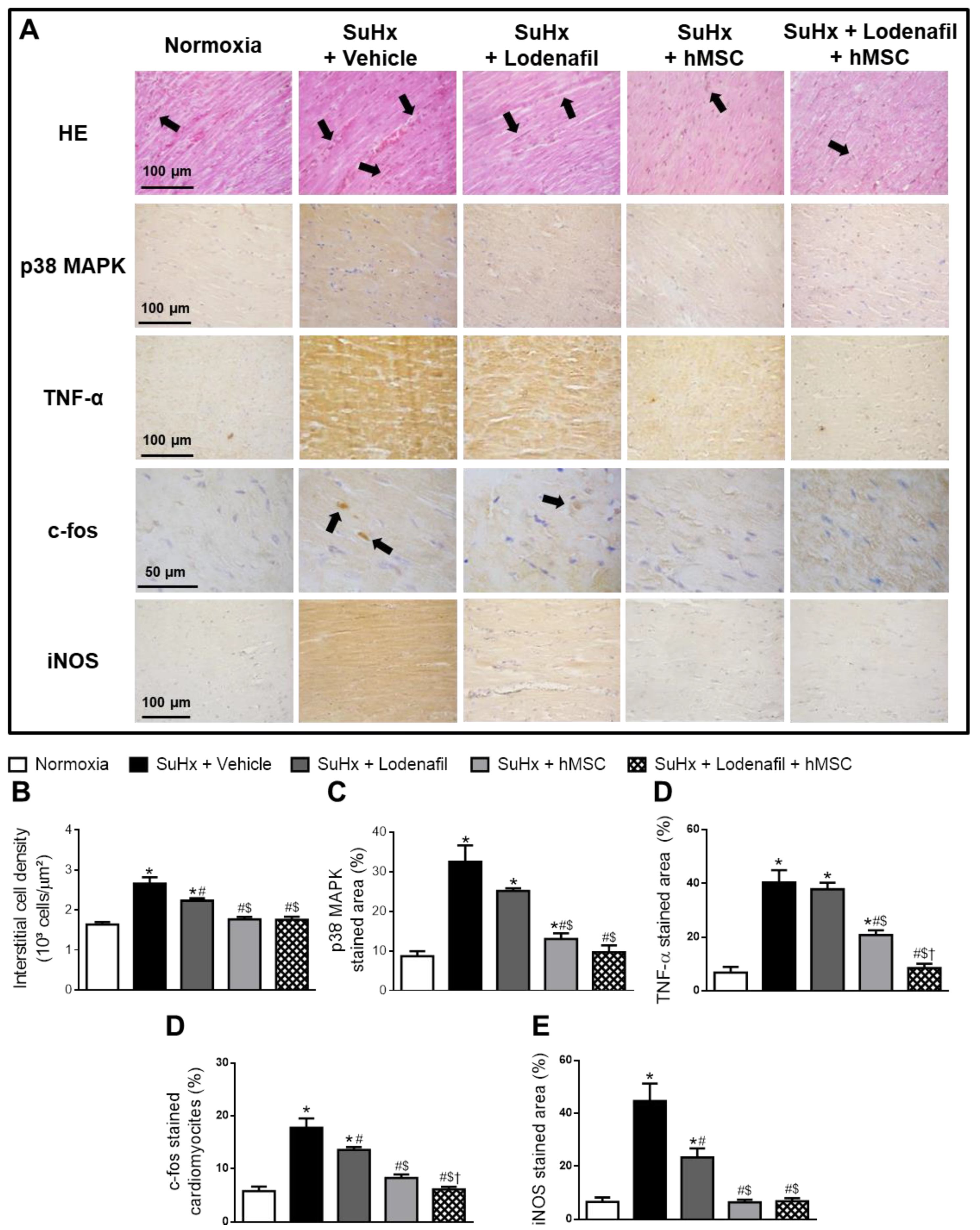
3.4. Lodenafil + hMSCs Therapy Reverses Increased Inflammatory Markers in the RV from SuHx-PAH
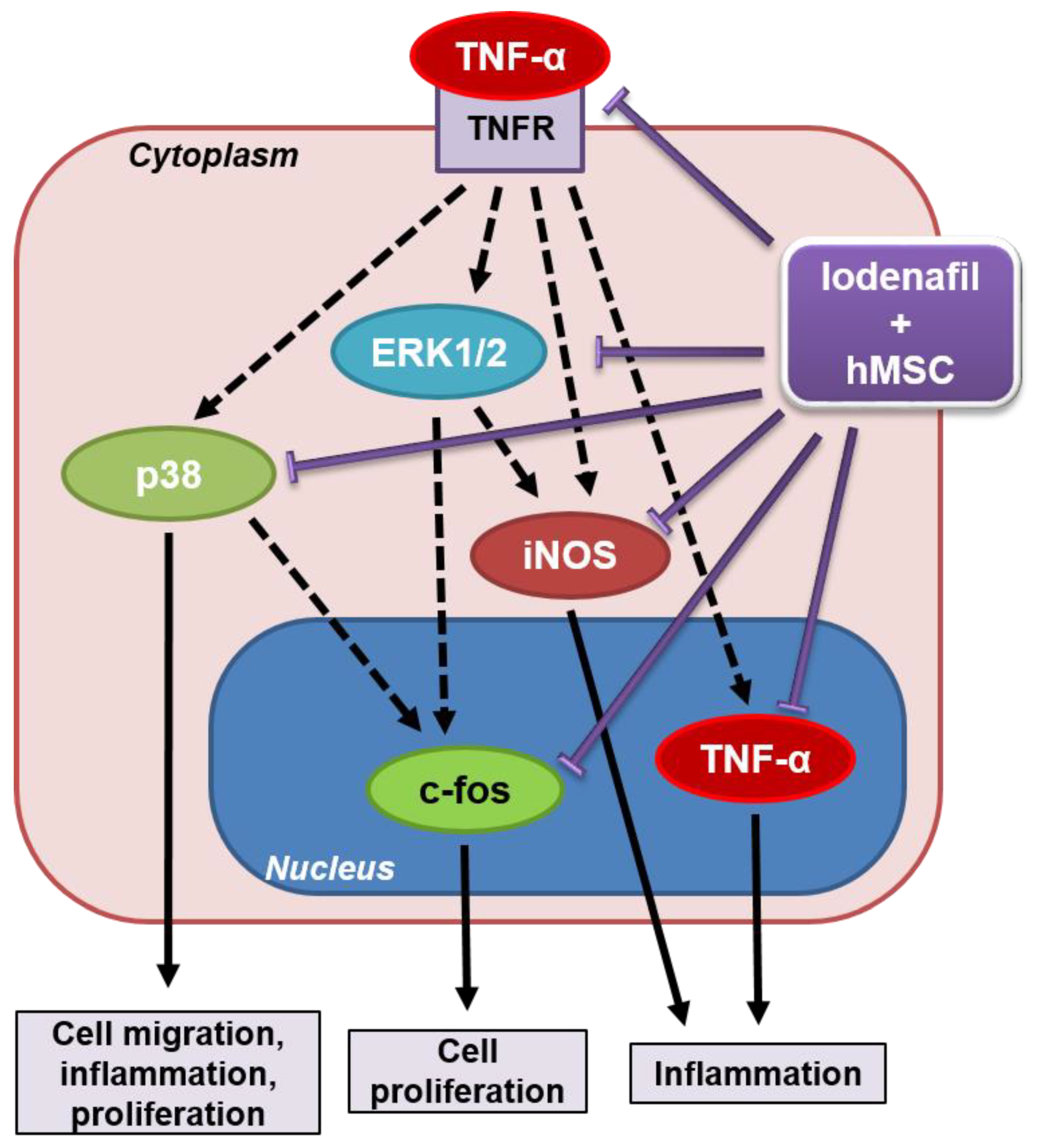
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Humbert, M.; Lau, E.M.T.; Montani, D.; Jaïs, X.; Sitbon, O.; Simmoneau, G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation 2014, 130, 2189–2208. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef]
- Guignabert, C.; Dorfmuller, P. Pathology and pathobiology of pulmonary hypertension. Semin. Respir. Crit. Care Med. 2013, 34, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.J.; Archer, S.L. The right ventricle in pulmonary arterial hypertension: Disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 2014, 115, 176–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galiè, N.; Channick, R.N.; Frantz, R.P.; Grünig, E.; Jing, Z.C.; Moiseeva, O.; Preston, I.R.; Pulido, T.; Safdar, Z.; Tamura, Y.; et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801889. [Google Scholar] [CrossRef]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [CrossRef]
- Andersson, K.-E. PDE5 inhibitors—pharmacology and clinical applications 20 years after sildenafil discovery. Br. J. Pharmacol. 2018, 175, 2554–2565. [Google Scholar] [CrossRef] [Green Version]
- Toque, H.A.; Teixeira, C.E.; Lorenzetti, R.; Okuyama, C.E.; Antunes, E.; de Nucci, G. Pharmacological characterization of a novel phosphodiesterase type 5 (PDE5) inhibitor lodenafil carbonate on human and rabbit corpus cavernosum. Eur. J. Pharmacol. 2008, 591, 189–195. [Google Scholar] [CrossRef]
- Polonio, I.B.; Acencio, M.M.; Pazetti, R.; de Almeida, F.M.; da Silva, B.S.; Pereira, K.A.B.; Souza, R. Lodenafil treatment in the monocrotaline model of pulmonary hypertension in rats. J. Bras. Pneumol. 2014, 40, 421–424. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, F.I.S. Reversão da Hipertensão Pulmonar Induzida Pela Monocrotalina em Ratos Pelo Inibidor da Enzima Fosfodiesterase 5, Lodenafila. Master’s Thesis, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil, 2017. [Google Scholar]
- Hoeper, M.M.; Kramer, T.; Pan, Z.; Eichstaedt, C.A.; Spiesshoefer, J.; Benjamin, N.; Olsson, K.M.; Meyer, K.; Vizza, C.D.; Vonk-Noordegraaf, A.; et al. Mortality in pulmonary arterial hypertension: Prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur. Respir. J. 2017, 50, 1700740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alencar, A.K.N.; Pimentel-Coelho, P.M.; Montes, G.C.; Silva, M.M.C.; Mendes, L.V.P.; Montagnoli, T.L.; Silva, A.M.S.; Vasques, J.F.; Rosado-de-Castro, P.H.; Gutfilen, B.; et al. Human mesenchymal stem cell therapy reverses SU5416/hypoxia-induced pulmonary arterial hypertension in mice. Front. Pharmacol. 2018, 6, 1395. [Google Scholar] [CrossRef]
- Gnecchi, M.; Danieli, P.; Malpasso, G.; Ciuffreda, M.C. Paracrine mechanisms of mesenchymal stem cells in tissue repair. In Mesenchymal Stem Cells—Methods and Protocols; Gnecchi, M., Ed.; Humana Press: New York, NY, USA, 2016; Volume 1416, pp. 123–146. ISBN 978-1-4939-3584-0. [Google Scholar]
- Fukumitsu, M.; Suzuki, K. Mesenchymal stem/stromal cell therapy for pulmonary arterial hypertension: Comprehensive review of preclinical studies. J. Cardiol. 2019, 74, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Schmuck, E.G.; Hacker, T.A.; Schreier, D.A.; Chesler, N.C.; Wang, Z. Beneficial effects of mesenchymal stem cell delivery via a novel cardiac bioscaffold on right ventricles of pulmonary arterial hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1005–H1013. [Google Scholar] [CrossRef]
- Klinger, J.R.; Pereira, M.; Del Tatto, M.; Brodsky, A.S.; Wu, K.Q.; Dooner, M.S.; Borgovan, T.; Wen, S.; Goldberg, L.R.; Aliotta, J.M.; et al. Mesenchymal stem cell extracellular vesicles reverse Sugen/hypoxia pulmonary hypertension in rats. Am. J. Respir. Cell Mol. Biol. 2020, 62, 577–587. [Google Scholar] [CrossRef]
- Bartolucci, J.; Verdugo, F.J.; González, P.L.; Larrea, R.E.; Abarzua, E.; Goset, C.; Rojo, P.; Palma, I.; Lamich, R.; Pedreros, P.A.; et al. Safety and efficacy of the intravenous infusion of umbilical cord mesenchymal stem cells in patients with heart failure—a phase 1/2 randomized controlled trial (RIMECARD trial [Randomized Clinical Trial of Intravenous Infusion Umbilical Cord Mesenchymal Stem Cells on Cardiopathy]). Circ. Res. 2017, 121, 1192–1204. [Google Scholar] [CrossRef]
- Sun, C.K.; Lin, Y.C.; Yuen, C.M.; Chua, S.; Chang, L.T.; Sheu, J.-J.; Lee, F.-Y.; Fu, M.; Leu, S.; Yip, H.-K. Enhanced protection against pulmonary hypertension with sildenafil and endothelial progenitor cell in rats. Int. J. Cardiol. 2012, 162, 45–58. [Google Scholar] [CrossRef]
- Yen, C.-H.; Tsai, T.H.; Leu, S.; Chen, Y.-L.; Chang, L.-T.; Chai, H.-T.; Chung, S.-Y.; Chua, S.; Tsai, C.-Y.; Chang, H.-W.; et al. Sildenafil improves long-term effect of endothelial progenitor cell-based treatment for monocrotaline-induced rat pulmonary arterial hypertension. Cytotherapy 2013, 15, 209–223. [Google Scholar] [CrossRef]
- Vitali, S.H.; Hansmann, G.; Rose, C.; Fernandez-Gonzalez, A.; Scheid, A.; Mitsialis, S.A.; Kourembanas, S. The Sugen 5416/hypoxia mouse model of pulmonary hypertension revisited: Long-term follow-up. Pulm. Circ. 2014, 4, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Urboniene, D.; Haber, I.; Fang, Y.H.; Thenappan, T.; Archer, S.L. Validation of high-resolution echocardiography and magnetic resonance imaging vs. high-fidelity catheterization in experimental pulmonary hypertension. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2010, 299, L401–L412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alencar, A.K.N.; Carvalho, F.I.; Silva, A.M.; Martinez, S.T.; Calasans-Maia, J.A.; Fraga, C.M.; Barreiro, E.J.; Zapata-Sudo, G.; Sudo, R.T. Synergistic interaction between a PDE5 inhibitor (sildenafil) and a new adenosine A2A receptor agonist (LASSBio-1359) improves pulmonary hypertension in rats. PLoS ONE 2018, 13, e0195047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alencar, A.K.N.; Pereira, S.L.; da Silva, F.E.; Mendes, L.V.P.; Cunha, V.M.N.; Lima, L.M.; Montagnoli, T.L.; Caruso-Neves, C.; Ferraz, E.B.; Tesch, R.; et al. N-acylhydrazone derivative ameliorates monocrotaline-induced pulmonary hypertension through the modulation of adenosine AA2R activity. Int. J. Cardiol. 2014, 173, 154–162. [Google Scholar] [CrossRef]
- Mello, T.G.; Rosado-de-Castro, P.H.; Campos, R.M.P.; Vasques, J.F.; Rangel-Junior, W.S.; Mattos, R.S.A.R.; Puig-Pijuan, T.; Foerster, B.U.; Gutfilen, B.; Souza, S.A.L.; et al. Intravenous human umbilical cord-derived mesenchymal stromal cell administration in models of moderate and severe intracerebral hemorrhage. Stem. Cells Dev. 2020, 29, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Furlani, D.; Ugurlucan, M.; Ong, L.-L. Is the intravascular administration of mesenchymal stem cells safe? Mesenchymal stem cells and intravital microscopy. Microvasc. Res. 2009, 77, 370–376. [Google Scholar] [CrossRef]
- Luan, Y.; Zhang, X.; Qi, T.-G.; Cheng, G.-H.; Sun, C.; Kong, F. Long-term research of stem cells in monocrotaline-induced pulmonary arterial hypertension. Clin. Exp. Med. 2013, 14, 439–446. [Google Scholar] [CrossRef]
- Zungu-Edmondson, M.; Shults, N.V.; Melnyk, O.; Suzuki, Y.J. Natural reversal of pulmonary vascular remodeling and right ventricular remodeling in SU5416/hypoxia-treated Sprague-Dawley rats. PLoS ONE 2017, 12, e0182551. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Yang, H.; Yue, H.; Strappe, P.M.; Xia, P.; Pan, L.; Zhang, Y.; Chai, S.; Chen, S.; Ma, L.; et al. Mesenchymal stem cells expressing eNOS and a Cav1 mutant inhibit vascular smooth muscle cell proliferation in a rat model of pulmonary hypertension. Heart Lung Circ. 2017, 26, 509–518. [Google Scholar] [CrossRef]
- Liang, X.; Ding, Y.; Zhang, Y.; Tse, H.-F.; Lian, Q. Paracrine mechanisms of mesenchymal stem cell-based therapy: Current status and perspectives. Cell Transplant. 2014, 23, 1045–1059. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Mitsialis, S.A.; Aslam, M.; Vitali, S.H.; Vergadi, E.; Konstantinou, G.; Sdrimas, K.; Fernandez-Gonzalez, A.; Kourembanas, S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation 2012, 126, 2601–2611. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Wang, X.; Li, Y.; He, L. Let-7a-transfected mesenchymal stem cells ameliorate monocrotaline-induced pulmonary hypertension by suppressing pulmonary artery smooth muscle cell growth through STAT3-BMPR2 signaling. Stem. Cell Res. Ther. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendonça, L.; Felix, N.S.; Blanco, N.G.; Silva, J.S.; Ferreira, T.P.; Abreu, S.C.; Cruz, F.F.; Rocha, N.; Silva, P.M.; Martins, V.; et al. Mesenchymal stromal cell therapy reduces lung inflammation and vascular remodeling and improves hemodynamics in experimental pulmonary arterial hypertension. Stem. Cell Res. Ther. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biasin, V.; Chwalek, K.; Wilhelm, J.; Best, J.; Marsh, L.M.; Ghanim, B.; Klepetko, W.; Fink, L.; Schermuly, R.T.; Weissmann, N.; et al. Endothelin-1 driven proliferation of pulmonary arterial smooth muscle cells is c-fos dependent. Int. J. Biochem. Cell Biol. 2014, 54, 137–148. [Google Scholar] [CrossRef]
- Xing, X.-Q.; Li, B.; Xu, S.-L.; Liu, J.; Zhang, C.-F.; Yang, J. MicroRNA-214-3p regulates hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration by targeting ARHGEF12. Med. Sci. Monit. 2019, 25, 5738–5746. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Tu, L.; Dewachter, L.; Gore, B.; Fadel, E.; Dartevelle, P.; Simonneau, G.; Humbert, M.; Eddahibi, S.; Guignabert, C. Autocrine fibroblast growth factor-2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2011, 45, 311–322. [Google Scholar] [CrossRef]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Kojonazarov, B.; Novoyatleva, T.; Boehm, M.; Happe, C.; Sibinska, Z.; Tian, X.; Sajjad, A.; Luitel, H.; Kriechling, P.; Posern, G.; et al. p38 MAPK inhibition improves heart function in pressure-loaded right ventricular hypertrophy. Am. J. Respir. Cell Mol. Biol. 2017, 57, 603–614. [Google Scholar] [CrossRef]
- Vanderpool, R.R.; Tang, H.; Rischard, F.; Yuan, J.X. Is p38 MAPK a dark force in right ventricular hypertrophy and failure in pulmonary arterial hypertension? Am. J. Respir. Cell Mol. Biol. 2017, 57, 506–508. [Google Scholar] [CrossRef]
- Itoh, T.; Nagaya, N.; Ishibashi-Ueda, H.; Kyotani, S.; Oya, H.; Sakamaki, F.; Kimura, H.; Nakanishi, N. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology 2006, 11, 158–163. [Google Scholar] [CrossRef]
- Urschel, K.; Cicha, I. TNF-α in the cardiovascular system: From physiology to therapy. Int. J. Interferon Cytokine Mediat. Res. 2015, 7, 9–25. [Google Scholar] [CrossRef] [Green Version]
- Leopold, J.A.; Maron, B.A. Molecular mechanisms of pulmonary vascular remodeling in pulmonary arterial hypertension. Int. J. Mol. Sci. 2016, 17, E761. [Google Scholar] [CrossRef]
- Dewatcher, L.; Dewatcher, C. Inflammation in right ventricular failure: Does it matter? Front Physiol. 2018, 9, 1056. [Google Scholar] [CrossRef]
- Cheng, T.-H.; Shih, N.-L.; Chen, S.-Y.; Lin, J.-W.; Chen, Y.-L.; Chen, C.-H.; Lin, H.; Cheng, C.-F.; Chiu, W.-T.; Wang, D.W.; et al. Nitric oxide inhibits endothelin-1-induced cardiomyocyte hypertrophy through cGMP-mediated suppression of extracellular-signal regulated kinase phosphorylation. Mol. Pharmacol. 2005, 68, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- Kuc, R.E.; Carlebur, M.; Maguire, J.J.; Yang, P.; Long, L.; Toshner, M.; Morrell, N.W.; Davenport, A.P. Modulation of endothelin receptors in the failing right ventricle of the heart and vasculature of the lung in human pulmonary arterial hypertension. Life Sci. 2014, 118, 391–396. [Google Scholar] [CrossRef] [Green Version]
- Muller, D.N.; Dechend, R.; Mervaala, E.M.A.; Park, J.-K.; Schmidt, F.; Fiebeler, A.; Theuer, J.; Breu, V.; Ganten, D.; Haller, H.; et al. NF-κB inhibition ameliorates angiotensin II–induced inflammatory damage in rats. Hypertension 2000, 35, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Bogaard, H.J.; Abe, K.; Noordegraaf, A.V.; Voelkel, N.F. The right ventricle under pressure: Cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009, 135, 794–804. [Google Scholar] [CrossRef] [Green Version]
- Nogueira-Ferreira, R.; Vitorino, R.; Ferreira, R.; Henriques-Coelho, T. Exploring the monocrotaline animal model for the study of pulmonary arterial hypertension: A network approach. Pulm. Pharmacol. Ther. 2015, 35, 8–16. [Google Scholar] [CrossRef]
- Imoto, K.; Okada, M.; Yamawaki, H. Periostin mediates right ventricular failure through induction of inducible nitric oxide synthase expression in right ventricular fibroblasts from monocrotaline-induced pulmonary arterial hypertensive rats. Int. J. Mol. Sci. 2019, 20, 62. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, M.d.M.C.d.; Alencar, A.K.N.d.; Silva, J.S.d.; Montagnoli, T.L.; Silva, G.F.d.; Rocha, B.d.S.; Montes, G.C.; Mendez-Otero, R.; Pimentel-Coelho, P.M.; Vasques, J.F.; et al. Therapeutic Benefit of the Association of Lodenafil with Mesenchymal Stem Cells on Hypoxia-induced Pulmonary Hypertension in Rats. Cells 2020, 9, 2120. https://doi.org/10.3390/cells9092120
Silva MdMCd, Alencar AKNd, Silva JSd, Montagnoli TL, Silva GFd, Rocha BdS, Montes GC, Mendez-Otero R, Pimentel-Coelho PM, Vasques JF, et al. Therapeutic Benefit of the Association of Lodenafil with Mesenchymal Stem Cells on Hypoxia-induced Pulmonary Hypertension in Rats. Cells. 2020; 9(9):2120. https://doi.org/10.3390/cells9092120
Chicago/Turabian StyleSilva, Marina de Moraes Carvalho da, Allan Kardec Nogueira de Alencar, Jaqueline Soares da Silva, Tadeu Lima Montagnoli, Grazielle Fernandes da Silva, Bruna de Souza Rocha, Guilherme Carneiro Montes, Rosália Mendez-Otero, Pedro Moreno Pimentel-Coelho, Juliana F. Vasques, and et al. 2020. "Therapeutic Benefit of the Association of Lodenafil with Mesenchymal Stem Cells on Hypoxia-induced Pulmonary Hypertension in Rats" Cells 9, no. 9: 2120. https://doi.org/10.3390/cells9092120