Hedgehog Signaling and Truncated GLI1 in Cancer


Abstract
:1. Introduction
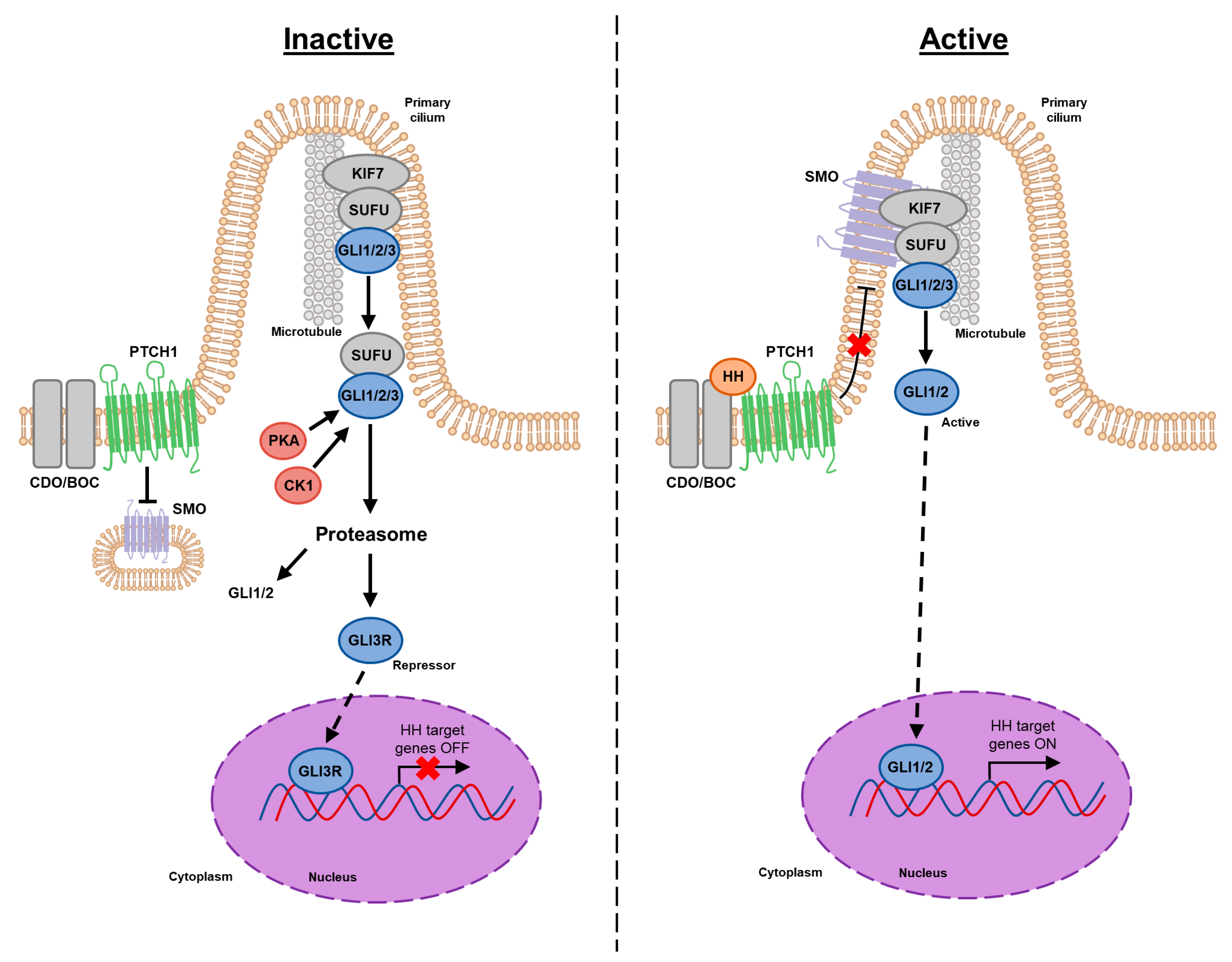
2. Canonical HH Signaling
3. Noncanonical Signaling
3.1. Type I—PTCH1 Functions Distinct from SMO Inhibition
3.2. Type II—SMO-Mediated Functions Independent of GLI1
3.3. Type III—Mechanisms Independent of Upstream PTCH1-SMO Signaling
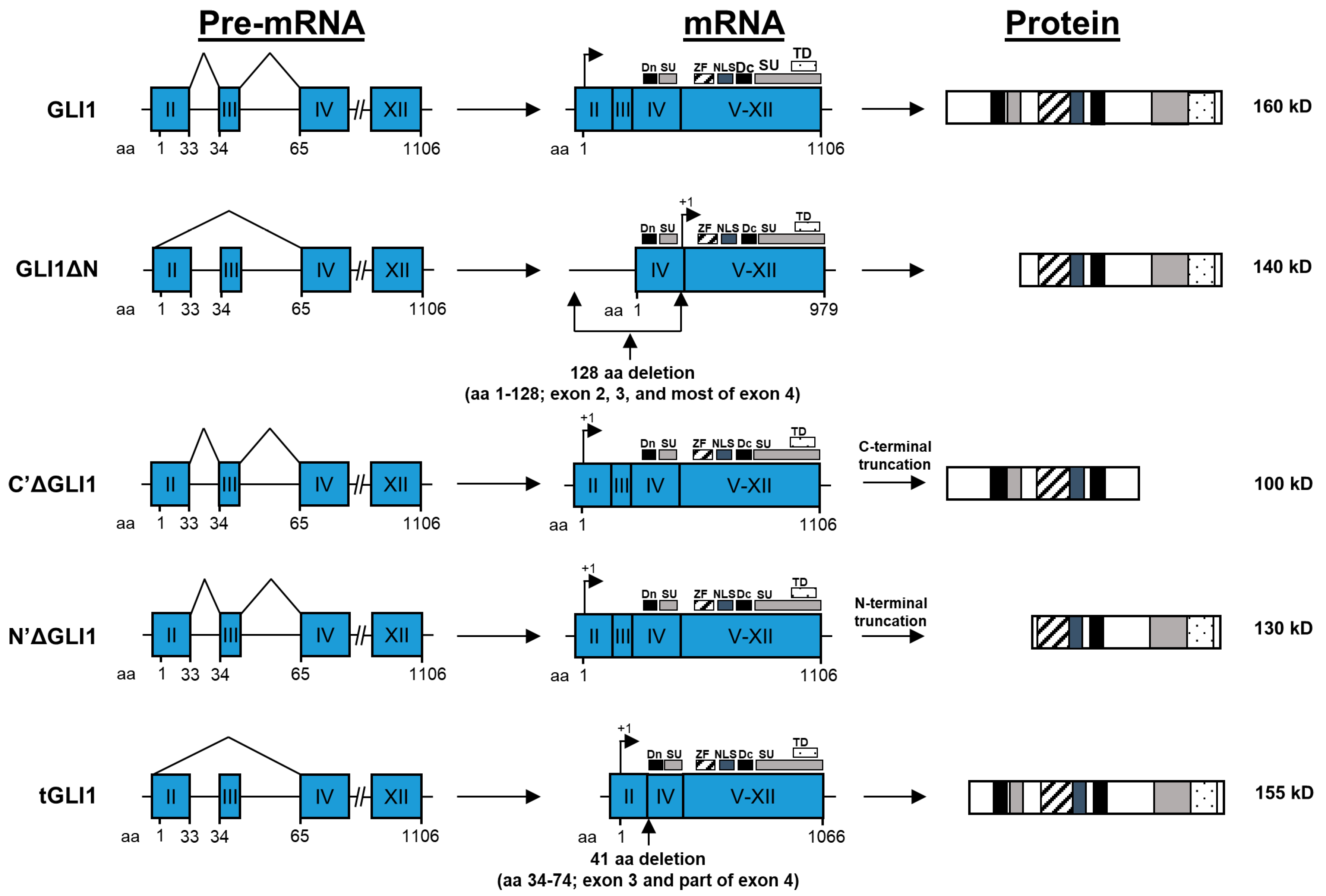
4. GLI1 Isoforms: Structures and Properties
4.1. GLI1
4.2. GLIΔN
4.3. C’ΔGLI1 and N’ΔGLI1
4.4. tGLI1
5. Aberrant SHH Signaling in Cancers
5.1. Type I Signaling—Autonomous and SHH Ligand-Independent
5.2. Type II Signaling—Autocrine/Juxtacrine SHH Ligand-Dependent
5.3. Type III Signaling—Paracrine SHH Ligand-Dependent
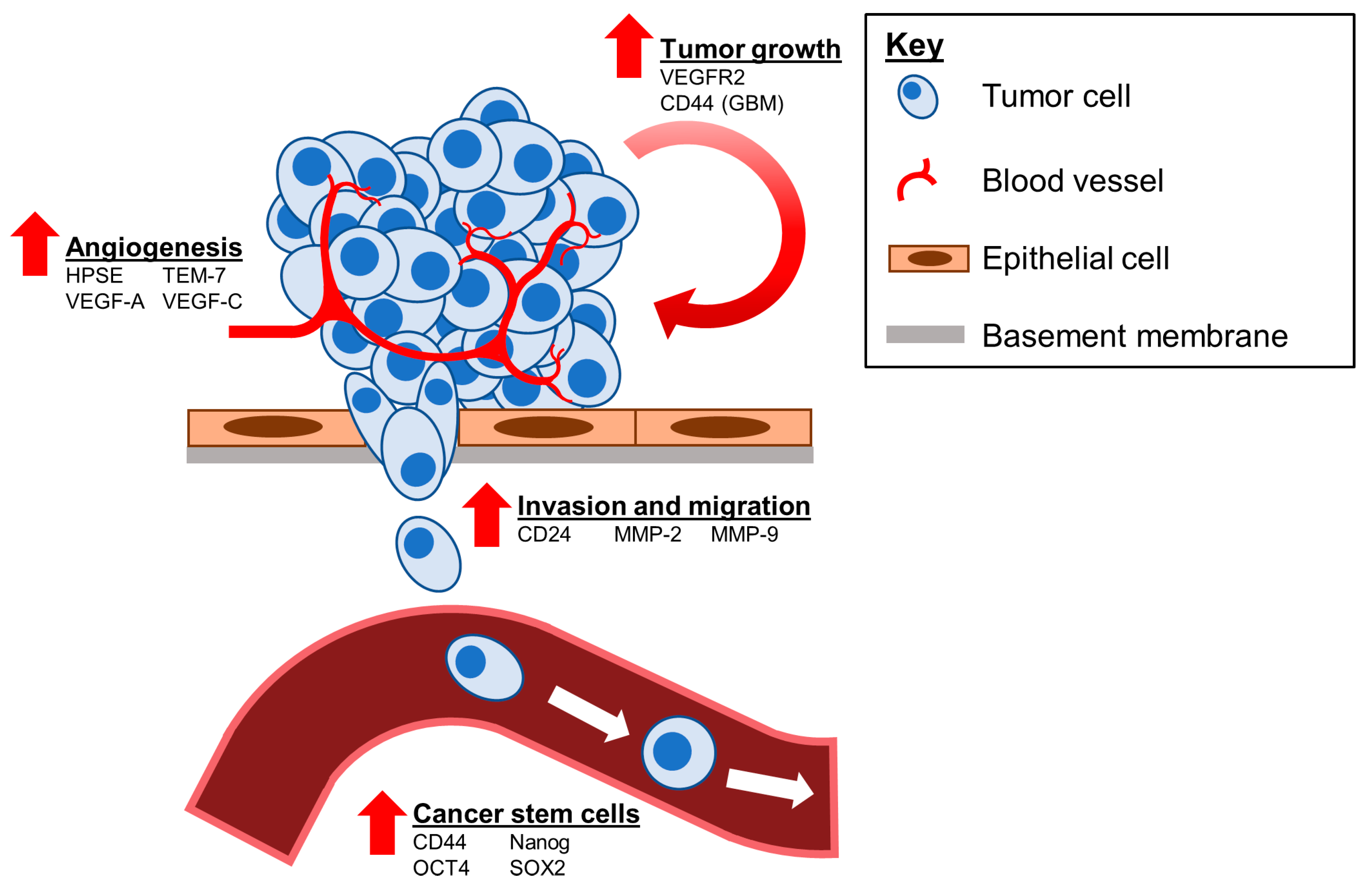
5.4. Cancer Stem Cells
6. SHH Signaling and Human Cancer
6.1. Glioblastoma
6.2. Breast Cancer
6.3. Therapeutic Targets Regulated by tGLI1
7. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef]
- Belloni, E.; Muenke, M.; Roessler, E.; Traverso, G.; Siegel-Bartelt, J.; Frumkin, A.; Mitchell, H.F.; Donis-Keller, H.; Helms, C.; Hing, A.V.; et al. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet. 1996, 14, 353–356. [Google Scholar] [CrossRef]
- Roessler, E.; Belloni, E.; Gaudenz, K.; Jay, P.; Berta, P.; Scherer, S.W.; Tsui, L.C.; Muenke, M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat. Genet. 1996, 14, 357–360. [Google Scholar] [CrossRef]
- Lum, L.; Beachy, P.A. The Hedgehog response network: Sensors, switches, and routers. Science 2004, 304, 1755–1759. [Google Scholar] [CrossRef] [Green Version]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [Green Version]
- Büller, N.V.J.A.; Rosekrans, S.L.; Westerlund, J.; van den Brink, G.R. Hedgehog Signaling and Maintenance of Homeostasis in the Intestinal Epithelium. Physiology 2012, 27, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Ihrie, R.A.; Shah, J.K.; Harwell, C.C.; Levine, J.H.; Guinto, C.D.; Lezameta, M.; Kriegstein, A.R.; Alvarez-Buylla, A. Persistent sonic hedgehog signaling in adult brain determines neural stem cell positional identity. Neuron 2011, 71, 250–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanas, G.; Benitah, S.A. Regenerating the skin: A task for the heterogeneous stem cell pool and surrounding niche. Nat. Rev. Mol. Cell Biol. 2013, 14, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Dahmane, N.; Lee, J.; Robins, P.; Heller, P.; Ruiz i Altaba, A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature 1997, 389, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Tostar, U.; Malm, C.J.; Meis-Kindblom, J.M.; Kindblom, L.G.; Toftgård, R.; Undén, A.B. Deregulation of the hedgehog signalling pathway: A possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J. Pathol. 2006, 208, 17–25. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Chang, Y.-C.; Kuo, Y.-L.; Lee, K.-T.; Chen, P.-S.; Cheung, C.H.A.; Chang, C.-P.; Phan, N.N.; Shen, M.-R.; Hsu, H.-P. Mutation of the PTCH1 gene predicts recurrence of breast cancer. Sci. Rep. 2019, 9, 16359. [Google Scholar] [CrossRef]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef]
- Sari, I.N.; Phi, L.T.H.; Jun, N.; Wijaya, Y.T.; Lee, S.; Kwon, H.Y. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells 2018, 7, 208. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, R.L.; Ray, H. Safety and Tolerability of Sonic Hedgehog Pathway Inhibitors in Cancer. Drug Saf. 2019, 42, 263–279. [Google Scholar] [CrossRef]
- Taipale, J.; Cooper, M.K.; Maiti, T.; Beachy, P.A. Patched acts catalytically to suppress the activity of Smoothened. Nature 2002, 418, 892–896. [Google Scholar] [CrossRef]
- Dunaeva, M.; Michelson, P.; Kogerman, P.; Toftgard, R. Characterization of the physical interaction of Gli proteins with SUFU proteins. J. Biol. Chem. 2003, 278, 5116–5122. [Google Scholar] [CrossRef] [Green Version]
- Ruiz i Altaba, A. Catching a Gli-mpse of Hedgehog. Cell 1997, 90, 193–196. [Google Scholar] [CrossRef] [Green Version]
- Shimokawa, T.; Tostar, U.; Lauth, M.; Palaniswamy, R.; Kasper, M.; Toftgård, R.; Zaphiropoulos, P.G. Novel human glioma-associated oncogene 1 (GLI1) splice variants reveal distinct mechanisms in the terminal transduction of the hedgehog signal. J. Biol. Chem. 2008, 283, 14345–14354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, H.W.; Zhu, H.; Cao, X.; Aldrich, A.; Ali-Osman, F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009, 69, 6790–6798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Geradts, J.; Dewhirst, M.W.; Lo, H.W. Upregulation of VEGF-A and CD24 gene expression by the tGLI1 transcription factor contributes to the aggressive behavior of breast cancer cells. Oncogene 2012, 31, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.H.; Ding, J.; Nguyen, S.; Liu, X.J.; Xu, G.; Zhou, H.Y.; Duan, N.N.; Yang, S.M.; Zern, M.A.; Wu, J. Aberrant hedgehog signaling is responsible for the highly invasive behavior of a subpopulation of hepatoma cells. Oncogene 2016, 35, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Gricius, D.; Kazlauskas, A. Analysis of oncogene GLI1 protein expression levels between differing grades of astrocytoma. Biologija 2014, 60, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Carpenter, R.L.; Lo, H.-W. TGLI1 Upregulates Expression of VEGFR2 and VEGF-A, Leading to a Robust VEGF-VEGFR2 Autocrine Loop and Cancer Cell Growth. Cancer Hallm. 2013, 1, 28–37. [Google Scholar] [CrossRef]
- Zhu, H.; Carpenter, R.L.; Han, W.; Lo, H.W. The GLI1 splice variant TGLI1 promotes glioblastoma angiogenesis and growth. Cancer Lett. 2014, 343, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, R.L.; Paw, I.; Zhu, H.; Sirkisoon, S.; Xing, F.; Watabe, K.; Debinski, W.; Lo, H.W. The gain-of-function GLI1 transcription factor TGLI1 enhances expression of VEGF-C and TEM7 to promote glioblastoma angiogenesis. Oncotarget 2015, 6, 22653–22665. [Google Scholar] [CrossRef] [Green Version]
- Rimkus, T.K.; Carpenter, R.L.; Sirkisoon, S.; Zhu, D.; Pasche, B.C.; Chan, M.D.; Lesser, G.J.; Tatter, S.B.; Watabe, K.; Debinski, W.; et al. Truncated Glioma-Associated Oncogene Homolog 1 (tGLI1) Mediates Mesenchymal Glioblastoma via Transcriptional Activation of CD44. Cancer Res. 2018, 78, 2589–2600. [Google Scholar] [CrossRef] [Green Version]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Anderson, A.; Harrison, A.; Lange, A.M.; Jin, G.; Watabe, K.; Lo, H.W. Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription factors promotes the aggressiveness of triple-negative breast cancers and HER2-enriched breast cancer. Oncogene 2018, 37, 2502–2514. [Google Scholar] [CrossRef] [PubMed]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Doheny, D.; Zhu, D.; Aguayo, N.R.; Xing, F.; Chan, M.; Ruiz, J.; Metheny-Barlow, L.J.; et al. TGLI1 transcription factor mediates breast cancer brain metastasis via activating metastasis-initiating cancer stem cells and astrocytes in the tumor microenvironment. Oncogene 2020, 39, 64–78. [Google Scholar] [CrossRef]
- Lee, J.; Ekker, S.; von Kessler, D.; Porter, J.; Sun, B.; Beachy, P. Autoproteolysis in hedgehog protein biogenesis. Science 1994, 266, 1528–1537. [Google Scholar] [CrossRef]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol Modification of Hedgehog Signaling Proteins in Animal Development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef]
- Pepinsky, R.B.; Zeng, C.; Wen, D.; Rayhorn, P.; Baker, D.P.; Williams, K.P.; Bixler, S.A.; Ambrose, C.M.; Garber, E.A.; Miatkowski, K.; et al. Identification of a palmitic acid-modified form of human Sonic hedgehog. J. Biol. Chem. 1998, 273, 14037–14045. [Google Scholar] [CrossRef] [Green Version]
- Buglino, J.A.; Resh, M.D. Hhat is a palmitoylacyltransferase with specificity for N-palmitoylation of Sonic Hedgehog. J. Biol. Chem. 2008, 283, 22076–22088. [Google Scholar] [CrossRef] [Green Version]
- Tenzen, T.; Allen, B.L.; Cole, F.; Kang, J.-S.; Krauss, R.S.; McMahon, A.P. The Cell Surface Membrane Proteins Cdo and Boc Are Components and Targets of the Hedgehog Signaling Pathway and Feedback Network in Mice. Dev. Cell 2006, 10, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Tukachinsky, H.; Petrov, K.; Watanabe, M.; Salic, A. Mechanism of inhibition of the tumor suppressor Patched by Sonic Hedgehog. Proc. Natl. Acad. Sci. USA 2016, 113, E5866–E5875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denef, N.; Neubüser, D.; Perez, L.; Cohen, S.M. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 2000, 102, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.R.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Cao, P.; Hu, M.; Gao, S.; Yan, N.; Gong, X. Inhibition of tetrameric Patched1 by Sonic Hedgehog through an asymmetric paradigm. Nat. Commun. 2019, 10, 2320. [Google Scholar] [CrossRef] [Green Version]
- Tseng, T.T.; Gratwick, K.S.; Kollman, J.; Park, D.; Nies, D.H.; Goffeau, A.; Saier, M.H., Jr. The RND permease superfamily: An ancient, ubiquitous and diverse family that includes human disease and development proteins. J. Mol. Microbiol. Biotechnol. 1999, 1, 107–125. [Google Scholar] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Incardona, J.P.; Gaffield, W.; Kapur, R.P.; Roelink, H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 1998, 125, 3553–3562. [Google Scholar]
- Bidet, M.; Joubert, O.; Lacombe, B.; Ciantar, M.; Nehmé, R.; Mollat, P.; Brétillon, L.; Faure, H.; Bittman, R.; Ruat, M.; et al. The hedgehog receptor patched is involved in cholesterol transport. PLoS ONE 2011, 6, e23834. [Google Scholar] [CrossRef]
- Doughty, D.M.; Coleman, M.L.; Hunter, R.C.; Sessions, A.L.; Summons, R.E.; Newman, D.K. The RND-family transporter, HpnN, is required for hopanoid localization to the outer membrane of Rhodopseudomonas palustris TIE-1. Proc. Natl. Acad. Sci. USA 2011, 108, E1045–E1051. [Google Scholar] [CrossRef] [Green Version]
- Bijlsma, M.F.; Spek, C.A.; Zivkovic, D.; van de Water, S.; Rezaee, F.; Peppelenbosch, M.P. Repression of smoothened by patched-dependent (pro-)vitamin D3 secretion. PLoS Biol. 2006, 4, e232. [Google Scholar] [CrossRef] [Green Version]
- Incardona, J.P. From sensing cellular sterols to assembling sensory structures. Dev. Cell 2005, 8, 798–799. [Google Scholar] [CrossRef] [Green Version]
- Kowatsch, C.; Woolley, R.E.; Kinnebrew, M.; Rohatgi, R.; Siebold, C. Structures of vertebrate Patched and Smoothened reveal intimate links between cholesterol and Hedgehog signalling. Curr. Opin. Struct. Biol. 2019, 57, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Undén, A.B.; Sandstedt, B.; Toftgård, R.; Zaphiropoulos, P.G. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat. Cell Biol. 1999, 1, 312–319. [Google Scholar] [CrossRef]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic Hedgehog dependent phosphorylation by CK1α and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, J.J.; Whalen, E.J.; Liu, R.; Xiao, K.; Kim, J.; Chen, M.; Wang, J.; Chen, W.; Lefkowitz, R.J. Beta-arrestin-mediated localization of smoothened to the primary cilium. Science 2008, 320, 1777–1781. [Google Scholar] [CrossRef] [Green Version]
- Milenkovic, L.; Scott, M.P.; Rohatgi, R. Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J. Cell Biol. 2009, 187, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.M.; Porter, J.A.; Chiang, C.; Chang, D.T.; Beachy, P.A.; Tessier-Lavigne, M. Long-range sclerotome induction by sonic hedgehog: Direct role of the amino-terminal cleavage product and modulation by the cyclic AMP signaling pathway. Cell 1995, 81, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Hammerschmidt, M.; Bitgood, M.J.; McMahon, A.P. Protein kinase A is a common negative regulator of Hedgehog signaling in the vertebrate embryo. Genes Dev. 1996, 10, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Roelink, H.; McKnight, G.S. Protein kinase A deficiency causes axially localized neural tube defects in mice. J. Biol. Chem. 2002, 277, 19889–19896. [Google Scholar] [CrossRef] [Green Version]
- Tuson, M.; He, M.; Anderson, K.V. Protein kinase A acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development 2011, 138, 4921–4930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [Green Version]
- Epstein, D.J.; Marti, E.; Scott, M.P.; McMahon, A.P. Antagonizing cAMP-dependent protein kinase A in the dorsal CNS activates a conserved Sonic hedgehog signaling pathway. Development 1996, 122, 2885–2894. [Google Scholar] [PubMed]
- Ruiz i Altaba, A. Gli proteins encode context-dependent positive and negative functions: Implications for development and disease. Development 1999, 126, 3205–3216. [Google Scholar]
- Kaesler, S.; Lüscher, B.; Rüther, U. Transcriptional activity of GLI1 is negatively regulated by protein kinase A. Biol. Chem. 2000, 381, 545–551. [Google Scholar] [CrossRef]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [Green Version]
- Liem, K.F.; He, M.; Ocbina, P.J.R.; Anderson, K.V. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 13377–13382. [Google Scholar] [CrossRef] [Green Version]
- Cheung, H.O.-L.; Zhang, X.; Ribeiro, A.; Mo, R.; Makino, S.; Puviindran, V.; Law, K.K.L.; Briscoe, J.; Hui, C.-C. The Kinesin Protein Kif7 Is a Critical Regulator of Gli Transcription Factors in Mammalian Hedgehog Signaling. Sci. Signal. 2009, 2, ra29. [Google Scholar] [CrossRef] [PubMed]
- Dorn, K.V.; Hughes, C.E.; Rohatgi, R. A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev. Cell 2012, 23, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Chen, W.; Chen, Y.; Jiang, J. Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res. 2012, 22, 1593–1604. [Google Scholar] [CrossRef]
- Han, Y.; Shi, Q.; Jiang, J. Multisite interaction with Sufu regulates Ci/Gli activity through distinct mechanisms in Hh signal transduction. Proc. Natl. Acad. Sci. USA 2015, 112, 6383–6388. [Google Scholar] [CrossRef] [Green Version]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varjosalo, M.; Taipale, J. Hedgehog signaling. J. Cell Sci. 2007, 120, 3–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, E.; De Smaele, E.; Di Marcotullio, L.; Screpanti, I.; Gulino, A. Hedgehog checkpoints in medulloblastoma: The chromosome 17p deletion paradigm. Trends Mol. Med. 2005, 11, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D. Hedgehog signalling: Emerging evidence for non-canonical pathways. Cell Signal 2009, 21, 1023–1034. [Google Scholar] [CrossRef]
- Brennan, D.; Chen, X.; Cheng, L.; Mahoney, M.; Riobo, N.A. Noncanonical Hedgehog signaling. Vitam. Horm. 2012, 88, 55–72. [Google Scholar] [CrossRef] [Green Version]
- Chinchilla, P.; Xiao, L.; Kazanietz, M.G.; Riobo, N.A. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle 2010, 9, 570–579. [Google Scholar] [CrossRef]
- Thibert, C.; Teillet, M.A.; Lapointe, F.; Mazelin, L.; Le Douarin, N.M.; Mehlen, P. Inhibition of neuroepithelial patched-induced apoptosis by sonic hedgehog. Science 2003, 301, 843–846. [Google Scholar] [CrossRef]
- Bredesen, D.E.; Mehlen, P.; Rabizadeh, S. Apoptosis and dependence receptors: A molecular basis for cellular addiction. Physiol. Rev. 2004, 84, 411–430. [Google Scholar] [CrossRef]
- Lu, X.; Liu, S.; Kornberg, T.B. The C-terminal tail of the Hedgehog receptor Patched regulates both localization and turnover. Genes Dev. 2006, 20, 2539–2551. [Google Scholar] [CrossRef] [Green Version]
- Nieuwenhuis, E.; Barnfield, P.C.; Makino, S.; Hui, C.C. Epidermal hyperplasia and expansion of the interfollicular stem cell compartment in mutant mice with a C-terminal truncation of Patched1. Dev. Biol. 2007, 308, 547–560. [Google Scholar] [CrossRef]
- Chang, H.; Li, Q.; Moraes, R.C.; Lewis, M.T.; Hamel, P.A. Activation of Erk by sonic hedgehog independent of canonical hedgehog signalling. Int. J. Biochem. Cell Biol. 2010, 42, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahmane, N.; Ruiz i Altaba, A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 1999, 126, 3089–3100. [Google Scholar] [PubMed]
- Wechsler-Reya, R.J.; Scott, M.P. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 1999, 22, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Roussel, M.F.; Hatten, M.E. Cerebellum development and medulloblastoma. Curr. Top. Dev. Biol. 2011, 94, 235–282. [Google Scholar] [CrossRef]
- Kenney, A.M.; Rowitch, D.H. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol. Cell. Biol. 2000, 20, 9055–9067. [Google Scholar] [CrossRef] [Green Version]
- Kenney, A.M.; Cole, M.D.; Rowitch, D.H. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 2003, 130, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Marak, B.N.; Dowarah, J.; Khiangte, L.; Singh, V.P. A comprehensive insight on the recent development of Cyclic Dependent Kinase inhibitors as anticancer agents. Eur. J. Med. Chem. 2020, 203, 112571. [Google Scholar] [CrossRef]
- Takizawa, C.G.; Morgan, D.O. Control of mitosis by changes in the subcellular location of cyclin-B1-Cdk1 and Cdc25C. Curr. Opin. Cell Biol. 2000, 12, 658–665. [Google Scholar] [CrossRef]
- Barnes, E.A.; Kong, M.; Ollendorff, V.; Donoghue, D.J. Patched1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J. 2001, 20, 2214–2223. [Google Scholar] [CrossRef] [Green Version]
- Riobo, N.A.; Saucy, B.; Dilizio, C.; Manning, D.R. Activation of heterotrimeric G proteins by Smoothened. Proc. Natl. Acad. Sci. USA 2006, 103, 12607–12612. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.; Nobes, C.D. Rho GTPases: Molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2000, 355, 965–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polizio, A.H.; Chinchilla, P.; Chen, X.; Kim, S.; Manning, D.R.; Riobo, N.A. Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J. Biol. Chem. 2011, 286, 19589–19596. [Google Scholar] [CrossRef] [Green Version]
- Ho Wei, L.; Arastoo, M.; Georgiou, I.; Manning, D.R.; Riobo-Del Galdo, N.A. Activation of the Gi protein-RHOA axis by non-canonical Hedgehog signaling is independent of primary cilia. PLoS ONE 2018, 13, e0203170. [Google Scholar] [CrossRef]
- Didiasova, M.; Schaefer, L.; Wygrecka, M. Targeting GLI Transcription Factors in Cancer. Molecules 2018, 23, 1003. [Google Scholar] [CrossRef] [Green Version]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Li, S.; Li, S.; Jiang, A.; Chen, Y.; Jiang, J. Hedgehog-induced phosphorylation by CK1 sustains the activity of Ci/Gli activator. Proc. Natl. Acad. Sci. USA 2014, 111, E5651–E5660. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Maye, P.; Kogerman, P.; Tejedor, F.J.; Toftgard, R.; Xie, W.; Wu, G.; Wu, D. Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J. Biol. Chem. 2002, 277, 35156–35161. [Google Scholar] [CrossRef] [Green Version]
- Ehe, B.K.; Lamson, D.R.; Tarpley, M.; Onyenwoke, R.U.; Graves, L.M.; Williams, K.P. Identification of a DYRK1A-mediated phosphorylation site within the nuclear localization sequence of the hedgehog transcription factor GLI1. Biochem. Biophys. Res. Commun. 2017, 491, 767–772. [Google Scholar] [CrossRef]
- Schneider, P.; Bayo-Fina, J.M.; Singh, R.; Kumar Dhanyamraju, P.; Holz, P.; Baier, A.; Fendrich, V.; Ramaswamy, A.; Baumeister, S.; Martinez, E.D.; et al. Identification of a novel actin-dependent signal transducing module allows for the targeted degradation of GLI1. Nat. Commun. 2015, 6, 8023. [Google Scholar] [CrossRef] [Green Version]
- Maloverjan, A.; Piirsoo, M.; Michelson, P.; Kogerman, P.; Osterlund, T. Identification of a novel serine/threonine kinase ULK3 as a positive regulator of Hedgehog pathway. Exp. Cell Res. 2010, 316, 627–637. [Google Scholar] [CrossRef]
- Maloverjan, A.; Piirsoo, M.; Kasak, L.; Peil, L.; Østerlund, T.; Kogerman, P. Dual function of UNC-51-like kinase 3 (Ulk3) in the Sonic hedgehog signaling pathway. J. Biol. Chem. 2010, 285, 30079–30090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [Green Version]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [Green Version]
- Mirza, A.N.; McKellar, S.A.; Urman, N.M.; Brown, A.S.; Hollmig, T.; Aasi, S.Z.; Oro, A.E. LAP2 Proteins Chaperone GLI1 Movement between the Lamina and Chromatin to Regulate Transcription. Cell 2019, 176, 198–212. [Google Scholar] [CrossRef] [Green Version]
- Mirza, A.N.; Fry, M.A.; Urman, N.M.; Atwood, S.X.; Roffey, J.; Ott, G.R.; Chen, B.; Lee, A.; Brown, A.S.; Aasi, S.Z.; et al. Combined inhibition of atypical PKC and histone deacetylase 1 is cooperative in basal cell carcinoma treatment. JCI Insight 2017, 2, e97071. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Luo, J.; Mosley, Y.Y.; Hedrick, V.E.; Paul, L.N.; Chang, J.; Zhang, G.; Wang, Y.K.; Banko, M.R.; Brunet, A.; et al. AMP-Activated Protein Kinase Directly Phosphorylates and Destabilizes Hedgehog Pathway Transcription Factor GLI1 in Medulloblastoma. Cell Rep. 2015, 12, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Di Magno, L.; Basile, A.; Coni, S.; Manni, S.; Sdruscia, G.; D’Amico, D.; Antonucci, L.; Infante, P.; De Smaele, E.; Cucchi, D.; et al. The energy sensor AMPK regulates Hedgehog signaling in human cells through a unique Gli1 metabolic checkpoint. Oncotarget 2016, 7, 9538–9549. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Huang, S.Y.; Ka-Wai Li, K.; Li, Y.H.; Hsu, W.H.; Zhang, G.J.; Chang, C.J.; Yang, J.Y. Dual degradation signals destruct GLI1: AMPK inhibits GLI1 through β-TrCP-mediated proteasome degradation. Oncotarget 2017, 8, 49869–49881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonucci, L.; Di Magno, L.; D’Amico, D.; Manni, S.; Serrao, S.M.; Di Pastena, F.; Bordone, R.; Yurtsever, Z.N.; Caimano, M.; Petroni, M.; et al. Mitogen-activated kinase kinase kinase 1 inhibits hedgehog signaling and medulloblastoma growth through GLI1 phosphorylation. Int. J. Oncol. 2019, 54, 505–514. [Google Scholar] [CrossRef]
- Shi, X.; Zhan, X.; Wu, J. A positive feedback loop between Gli1 and tyrosine kinase Hck amplifies shh signaling activities in medulloblastoma. Oncogenesis 2015, 4, e176. [Google Scholar] [CrossRef] [Green Version]
- Celebi, G.; Kesim, H.; Ozer, E.; Kutlu, O. The Effect of Dysfunctional Ubiquitin Enzymes in the Pathogenesis of Most Common Diseases. Int. J. Mol. Sci. 2020, 21, 6335. [Google Scholar] [CrossRef]
- Deng, W.; Vanderbilt, D.B.; Lin, C.C.; Martin, K.H.; Brundage, K.M.; Ruppert, J.M. SOX9 inhibits β-TrCP-mediated protein degradation to promote nuclear GLI1 expression and cancer stem cell properties. J. Cell Sci. 2015, 128, 1123–1138. [Google Scholar] [CrossRef] [Green Version]
- Huntzicker, E.G.; Estay, I.S.; Zhen, H.; Lokteva, L.A.; Jackson, P.K.; Oro, A.E. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006, 20, 276–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marcotullio, L.; Greco, A.; Mazzà, D.; Canettieri, G.; Pietrosanti, L.; Infante, P.; Coni, S.; Moretti, M.; De Smaele, E.; Ferretti, E.; et al. Numb activates the E3 ligase Itch to control Gli1 function through a novel degradation signal. Oncogene 2011, 30, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Di Marcotullio, L.; Ferretti, E.; Greco, A.; De Smaele, E.; Po, A.; Sico, M.A.; Alimandi, M.; Giannini, G.; Maroder, M.; Screpanti, I.; et al. Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nat. Cell Biol. 2006, 8, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.; Briscoe, J.; Ulloa, F. SUMOylation by Pias1 regulates the activity of the Hedgehog dependent Gli transcription factors. PLoS ONE 2010, 5, e11996. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yan, S.; Ding, J.; Yu, T.T.; Cheng, S.Y. DeSUMOylation of Gli1 by SENP1 Attenuates Sonic Hedgehog Signaling. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Miele, E.; Po, A.; Begalli, F.; Antonucci, L.; Mastronuzzi, A.; Marras, C.E.; Carai, A.; Cucchi, D.; Abballe, L.; Besharat, Z.M.; et al. β-arrestin1-mediated acetylation of Gli1 regulates Hedgehog/Gli signaling and modulates self-renewal of SHH medulloblastoma cancer stem cells. BMC Cancer 2017, 17, 488. [Google Scholar] [CrossRef] [Green Version]
- Coni, S.; Mancuso, A.B.; Di Magno, L.; Sdruscia, G.; Manni, S.; Serrao, S.M.; Rotili, D.; Spiombi, E.; Bufalieri, F.; Petroni, M.; et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci. Rep. 2017, 7, 44079. [Google Scholar] [CrossRef]
- Das, S.; Bailey, S.K.; Metge, B.J.; Hanna, A.; Hinshaw, D.C.; Mota, M.; Forero-Torres, A.; Chatham, J.C.; Samant, R.S.; Shevde, L.A. O-GlcNAcylation of GLI transcription factors in hyperglycemic conditions augments Hedgehog activity. Lab. Investig. J. Tech. Methods Pathol. 2019, 99, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Bigner, S.H.; Bigner, D.D.; Trent, J.M.; Law, M.L.; O’Brien, S.J.; Wong, A.J.; Vogelstein, B. Identification of an amplified, highly expressed gene in a human glioma. Science 1987, 236, 70–73. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Ruppert, J.M.; Bigner, S.H.; Vogelstein, B. The GLI gene is a member of the Kruppel family of zinc finger proteins. Nature 1988, 332, 371–374. [Google Scholar] [CrossRef]
- Ruppert, J.M.; Kinzler, K.W.; Wong, A.J.; Bigner, S.H.; Kao, F.T.; Law, M.L.; Seuanez, H.N.; O’Brien, S.J.; Vogelstein, B. The GLI-Kruppel family of human genes. Mol. Cell. Biol. 1988, 8, 3104–3113. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell. Biol. 1990, 10, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Winklmayr, M.; Schmid, C.; Laner-Plamberger, S.; Kaser, A.; Aberger, F.; Eichberger, T.; Frischauf, A.-M. Non-consensus GLI binding sites in Hedgehog target gene regulation. BMC Mol. Biol. 2010, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Pavletich, N.P.; Pabo, C.O. Crystal structure of a five-finger GLI-DNA complex: New perspectives on zinc fingers. Science 1993, 261, 1701–1707. [Google Scholar] [CrossRef]
- Bauer, N.C.; Doetsch, P.W.; Corbett, A.H. Mechanisms Regulating Protein Localization. Traffic 2015, 16, 1039–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatayama, M.; Aruga, J. Gli protein nuclear localization signal. Vitam. Horm. 2012, 88, 73–89. [Google Scholar] [CrossRef]
- Malatesta, M.; Steinhauer, C.; Mohammad, F.; Pandey, D.P.; Squatrito, M.; Helin, K. Histone acetyltransferase PCAF is required for Hedgehog-Gli-dependent transcription and cancer cell proliferation. Cancer Res. 2013, 73, 6323–6333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagani, Z.; Mora-Blanco, E.L.; Sansam, C.G.; McKenna, E.S.; Wilson, B.; Chen, D.; Klekota, J.; Tamayo, P.; Nguyen, P.T.; Tolstorukov, M.; et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 2010, 16, 1429–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, X.; Shi, X.; Zhang, Z.; Chen, Y.; Wu, J.I. Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proc. Natl. Acad. Sci. USA 2011, 108, 12758–12763. [Google Scholar] [CrossRef] [Green Version]
- Roessler, E.; Ermilov, A.N.; Grange, D.K.; Wang, A.; Grachtchouk, M.; Dlugosz, A.A.; Muenke, M. A previously unidentified amino-terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum. Mol. Genet. 2005, 14, 2181–2188. [Google Scholar] [CrossRef] [Green Version]
- Speek, M.; Njunkova, O.; Pata, I.; Valdre, E.; Kogerman, P. A potential role of alternative splicing in the regulation of the transcriptional activity of human GLI2 in gonadal tissues. BMC Mol. Biol. 2006, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsanev, R.; Tiigimägi, P.; Michelson, P.; Metsis, M.; Østerlund, T.; Kogerman, P. Identification of the gene transcription repressor domain of Gli3. FEBS Lett. 2009, 583, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Stecca, B.; Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Tostar, U.; Finta, C.; Rahman, M.F.; Shimokawa, T.; Zaphiropoulos, P.G. Novel mechanism of action on Hedgehog signaling by a suppressor of fused carboxy terminal variant. PLoS ONE 2012, 7, e37761. [Google Scholar] [CrossRef] [Green Version]
- Piirsoo, A.; Pink, A.; Kasak, L.; Kala, M.; Kasvandik, S.; Ustav, M.; Piirsoo, M. Differential phosphorylation determines the repressor and activator potencies of GLI1 proteins and their efficiency in modulating the HPV life cycle. PLoS ONE 2019, 14, e0225775. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.R.; Kunkalla, K.; Qu, C.; Schlette, E.; Neelapu, S.S.; Samaniego, F.; Vega, F. ABCG2 is a direct transcriptional target of hedgehog signaling and involved in stroma-induced drug tolerance in diffuse large B-cell lymphoma. Oncogene 2011, 30, 4874–4886. [Google Scholar] [CrossRef] [Green Version]
- Tolosa, E.; Fernandez-Barrena, M.G.; Iguchi, E.; McCleary-Wheeler, A.; Carr, R.; Almada, L.; Flores, L.; Vera, R.E.; Alfonse, G.; Marks, D.; et al. GLI1/GLI2 functional interplay is required to control Hedgehog/GLI targets gene expression. Biochem. J. 2020, 477, 3131–3145. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.Q.; Ye, M.; Yu, P.G.; Xiao, C.; Lin, F.Y. Glioma-Associated Oncogene Homolog1 (Gli1)-Aquaporin1 pathway promotes glioma cell metastasis. BMB Rep. 2016, 49, 394–399. [Google Scholar] [CrossRef] [Green Version]
- Mazumdar, T.; DeVecchio, J.; Shi, T.; Jones, J.; Agyeman, A.; Houghton, J.A. Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res. 2011, 71, 1092–1102. [Google Scholar] [CrossRef] [Green Version]
- Bigelow, R.L.; Chari, N.S.; Unden, A.B.; Spurgers, K.B.; Lee, S.; Roop, D.R.; Toftgard, R.; McDonnell, T.J. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. J. Biol. Chem. 2004, 279, 1197–1205. [Google Scholar] [CrossRef] [Green Version]
- Hegde, G.V.; Peterson, K.J.; Emanuel, K.; Mittal, A.K.; Joshi, A.D.; Dickinson, J.D.; Kollessery, G.J.; Bociek, R.G.; Bierman, P.; Vose, J.M.; et al. Hedgehog-induced survival of B-cell chronic lymphocytic leukemia cells in a stromal cell microenvironment: A potential new therapeutic target. Mol. Cancer Res. 2008, 6, 1928–1936. [Google Scholar] [CrossRef] [Green Version]
- Inaguma, S.; Riku, M.; Hashimoto, M.; Murakami, H.; Saga, S.; Ikeda, H.; Kasai, K. GLI1 Interferes with the DNA Mismatch Repair System in Pancreatic Cancer through BHLHE41-Mediated Suppression of MLH1. Cancer Res. 2013, 73, 7313–7323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Venugopal, C.; Manoranjan, B.; McFarlane, N.; O’Farrell, E.; Nolte, S.; Gunnarsson, T.; Hollenberg, R.; Kwiecien, J.; Northcott, P.; et al. Sonic hedgehog regulates Bmi1 in human medulloblastoma brain tumor-initiating cells. Oncogene 2012, 31, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Nanta, R.; Sharma, J.; Gunewardena, S.; Singh, K.P.; Shankar, S.; Srivastava, R.K. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget 2015, 6, 32039–32060. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Riku, M.; Ito, H.; Tsunoda, T.; Ikeda, H.; Kasai, K. GLI1 orchestrates CXCR4/CXCR7 signaling to enhance migration and metastasis of breast cancer cells. Oncotarget 2015, 6, 33648–33657. [Google Scholar] [CrossRef] [Green Version]
- Eberl, M.; Klingler, S.; Mangelberger, D.; Loipetzberger, A.; Damhofer, H.; Zoidl, K.; Schnidar, H.; Hache, H.; Bauer, H.C.; Solca, F.; et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol. Med. 2012, 4, 218–233. [Google Scholar] [CrossRef]
- Nye, M.D.; Almada, L.L.; Fernandez-Barrena, M.G.; Marks, D.L.; Elsawa, S.F.; Vrabel, A.; Tolosa, E.J.; Ellenrieder, V.; Fernandez-Zapico, M.E. The transcription factor GLI1 interacts with SMAD proteins to modulate transforming growth factor β-induced gene expression in a p300/CREB-binding protein-associated factor (PCAF)-dependent manner. J. Biol. Chem. 2014, 289, 15495–15506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.W.; Kita, Y.; Frank, D.J.; Majewski, R.R.; Konicek, B.A.; Nobrega, M.A.; Jacob, H.; Walterhouse, D.; Iannaccone, P. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J. Biol. Chem. 2002, 277, 5548–5555. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.W.; Gilbertson, R.; Iannaccone, S.; Iannaccone, P.; Walterhouse, D. Defining a role for Sonic hedgehog pathway activation in desmoplastic medulloblastoma by identifying GLI1 target genes. Int. J. Cancer 2009, 124, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahi, M.H.; Afzal, M.; Sinha, S.; Eberhart, C.G.; Rey, J.A.; Fan, X.; Castresana, J.S. Regulation of sonic hedgehog-GLI1 downstream target genes PTCH1, Cyclin D2, Plakoglobin, PAX6 and NKX2.2 and their epigenetic status in medulloblastoma and astrocytoma. BMC Cancer 2010, 10, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Wang, F.; Yang, L.; Guo, C.; Wan, R.; Ke, A.; Xu, L.; Hu, G.; Xu, X.; Shen, J.; et al. Expression of DNMT1 and DNMT3a are regulated by GLI1 in human pancreatic cancer. PLoS ONE 2011, 6, e27684. [Google Scholar] [CrossRef] [Green Version]
- Diao, Y.; Rahman, M.F.; Vyatkin, Y.; Azatyan, A.; St Laurent, G.; Kapranov, P.; Zaphiropoulos, P.G. Identification of novel GLI1 target genes and regulatory circuits in human cancer cells. Mol. Oncol. 2018, 12, 1718–1734. [Google Scholar] [CrossRef]
- Pandolfi, S.; Montagnani, V.; Lapucci, A.; Stecca, B. HEDGEHOG/GLI-E2F1 axis modulates iASPP expression and function and regulates melanoma cell growth. Cell Death Differ. 2015, 22, 2006–2019. [Google Scholar] [CrossRef]
- Pignot, G.; Vieillefond, A.; Vacher, S.; Zerbib, M.; Debre, B.; Lidereau, R.; Amsellem-Ouazana, D.; Bieche, I. Hedgehog pathway activation in human transitional cell carcinoma of the bladder. Br. J. Cancer 2012, 106, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Teh, M.T.; Wong, S.T.; Neill, G.W.; Ghali, L.R.; Philpott, M.P.; Quinn, A.G. FOXM1 is a downstream target of Gli1 in basal cell carcinomas. Cancer Res. 2002, 62, 4773–4780. [Google Scholar]
- Xue, J.; Zhou, A.; Tan, C.; Wu, Y.; Lee, H.T.; Li, W.; Xie, K.; Huang, S. Forkhead Box M1 Is Essential for Nuclear Localization of Glioma-associated Oncogene Homolog 1 in Glioblastoma Multiforme Cells by Promoting Importin-7 Expression. J. Biol. Chem. 2015, 290, 18662–18670. [Google Scholar] [CrossRef] [Green Version]
- Long, B.; Zhu, H.; Zhu, C.; Liu, T.; Meng, W. Activation of the Hedgehog pathway in chronic myelogeneous leukemia patients. J. Exp. Clin. Cancer Res. 2011, 30, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blotta, S.; Jakubikova, J.; Calimeri, T.; Roccaro, A.M.; Amodio, N.; Azab, A.K.; Foresta, U.; Mitsiades, C.S.; Rossi, M.; Todoerti, K.; et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood 2012, 120, 5002–5013. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Peng, Z.; Lin, J.; Ren, X.; Zhang, G.; Cui, Y. Forkhead box C1 boosts triple-negative breast cancer metastasis through activating the transcription of chemokine receptor-4. Cancer Sci. 2018, 109, 3794–3804. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Lo, H.W. The Human Glioma-Associated Oncogene Homolog 1 (GLI1) Family of Transcription Factors in Gene Regulation and Diseases. Curr. Genom. 2010, 11, 238–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnat, F.; Siegl-Cachedenier, I.; Malerba, M.; Gervaz, P.; Ruiz i Altaba, A. Loss of WNT-TCF addiction and enhancement of HH-GLI1 signalling define the metastatic transition of human colon carcinomas. EMBO Mol. Med. 2010, 2, 440–457. [Google Scholar] [CrossRef]
- Inaguma, S.; Kasai, K.; Ikeda, H. GLI1 facilitates the migration and invasion of pancreatic cancer cells through MUC5AC-mediated attenuation of E-cadherin. Oncogene 2011, 30, 714–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Po, A.; Ferretti, E.; Miele, E.; De Smaele, E.; Paganelli, A.; Canettieri, G.; Coni, S.; Di Marcotullio, L.; Biffoni, M.; Massimi, L.; et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J. 2010, 29, 2646–2658. [Google Scholar] [CrossRef] [Green Version]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.N.; Borges, I.; Ruiz i Altaba, A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef] [Green Version]
- Casas, B.S.; Adolphe, C.; Lois, P.; Navarrete, N.; Solís, N.; Bustamante, E.; Gac, P.; Cabané, P.; Gallegos, I.; Wainwright, B.J.; et al. Downregulation of the Sonic Hedgehog/Gli pathway transcriptional target Neogenin-1 is associated with basal cell carcinoma aggressiveness. Oncotarget 2017, 8, 84006–84018. [Google Scholar] [CrossRef] [Green Version]
- Milla, L.A.; Arros, A.; Espinoza, N.; Remke, M.; Kool, M.; Taylor, M.D.; Pfister, S.M.; Wainwright, B.J.; Palma, V. Neogenin1 is a sonic hedgehog target in medulloblastoma and is necessary for cell cycle progression. Int. J. Cancer 2014, 134, 21–31. [Google Scholar] [CrossRef]
- Xie, J.; Aszterbaum, M.; Zhang, X.; Bonifas, J.M.; Zachary, C.; Epstein, E.; McCormick, F. A role of PDGFRalpha in basal cell carcinoma proliferation. Proc. Natl. Acad. Sci. USA 2001, 98, 9255–9259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunarta, I.K.; Li, R.; Nakazato, R.; Suzuki, R.; Boldbaatar, J.; Suzuki, T.; Yoshioka, K. Critical role of glioma-associated oncogene homolog 1 in maintaining invasive and mesenchymal-like properties of melanoma cells. Cancer Sci. 2017, 108, 1602–1611. [Google Scholar] [CrossRef] [Green Version]
- Bora-Singhal, N.; Perumal, D.; Nguyen, J.; Chellappan, S. Gli1-Mediated Regulation of Sox2 Facilitates Self-Renewal of Stem-Like Cells and Confers Resistance to EGFR Inhibitors in Non-Small Cell Lung Cancer. Neoplasia 2015, 17, 538–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.; Carpenter, R.L.; Cao, X.; Lo, H.W. STAT1 gene expression is enhanced by nuclear EGFR and HER2 via cooperation with STAT3. Mol. Carcinog. 2013, 52, 959–969. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Zapico, M.E. GLI1 finds a new role in cancer stem cell biology. EMBO Mol. Med. 2013, 5, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef]
- Gorlin, R.J.; Goltz, R.W. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N. Engl. J. Med. 1960, 262, 908–912. [Google Scholar] [CrossRef]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef]
- Springate, J.E. The nevoid basal cell carcinoma syndrome. J. Pediatr. Surg. 1986, 21, 908–910. [Google Scholar] [CrossRef]
- Evans, D.G.R.; Farndon, P.A.; Burnell, L.D.; Gattamaneni, H.R.; Birch, J.M. The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br. J. Cancer 1991, 64, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Reifenberger, J.; Wolter, M.; Weber, R.G.; Megahed, M.; Ruzicka, T.; Lichter, P.; Reifenberger, G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998, 58, 1798–1803. [Google Scholar]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef]
- Lee, Y.; Miller, H.L.; Jensen, P.; Hernan, R.; Connelly, M.; Wetmore, C.; Zindy, F.; Roussel, M.F.; Curran, T.; Gilbertson, R.J.; et al. A Molecular Fingerprint for Medulloblastoma. Cancer Res. 2003, 63, 5428–5437. [Google Scholar]
- Ellison, D.W.; Dalton, J.; Kocak, M.; Nicholson, S.L.; Fraga, C.; Neale, G.; Kenney, A.M.; Brat, D.J.; Perry, A.; Yong, W.H.; et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011, 121, 381–396. [Google Scholar] [CrossRef] [Green Version]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Kohler, B.; Schonicke, A.; Scharwachter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Kimura, F.; Florl, A.R.; Seifert, H.H.; Louhelainen, J.; Maas, S.; Knowles, M.A.; Schulz, W.A. Destabilization of chromosome 9 in transitional cell carcinoma of the urinary bladder. Br. J. Cancer 2001, 85, 1887–1893. [Google Scholar] [CrossRef]
- Yang, L.; Wang, L.S.; Chen, X.L.; Gatalica, Z.; Qiu, S.; Liu, Z.; Stoner, G.; Zhang, H.; Weiss, H.; Xie, J. Hedgehog signaling activation in the development of squamous cell carcinoma and adenocarcinoma of esophagus. Int. J. Biochem. Mol. Biol. 2012, 3, 46–57. [Google Scholar] [PubMed]
- Saito, T.; Mitomi, H.; Imamhasan, A.; Hayashi, T.; Kurisaki-Arakawa, A.; Mitani, K.; Takahashi, M.; Kajiyama, Y.; Yao, T. PTCH1 mutation is a frequent event in oesophageal basaloid squamous cell carcinoma. Mutagenesis 2015, 30, 297–301. [Google Scholar] [CrossRef]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theunissen, J.W.; de Sauvage, F.J. Paracrine Hedgehog signaling in cancer. Cancer Res. 2009, 69, 6007–6010. [Google Scholar] [CrossRef] [Green Version]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [Green Version]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef]
- Fan, L.; Pepicelli, C.V.; Dibble, C.C.; Catbagan, W.; Zarycki, J.L.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M.L.; Munoz, A.; et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004, 145, 3961–3970. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Sheng, T.; Zhang, Y.; Zhang, X.; He, J.; Huang, S.; Chen, K.; Sultz, J.; Adegboyega, P.A.; Zhang, H.; et al. Hedgehog signaling is activated in subsets of esophageal cancers. Int. J. Cancer 2006, 118, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Aberger, F.; Hutterer, E.; Sternberg, C.; Del Burgo, P.J.; Hartmann, T.N. Acute myeloid leukemia - strategies and challenges for targeting oncogenic Hedgehog/GLI signaling. Cell Commun. Signal. 2017, 15, 8. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chen, F.; Zhu, Q.; Ding, B.; Zhong, Q.; Huang, K.; Jiang, X.; Wang, Z.; Yin, C.; Zhu, Y.; et al. Gli-1/PI3K/AKT/NF-kB pathway mediates resistance to radiation and is a target for reversion of responses in refractory acute myeloid leukemia cells. Oncotarget 2016, 7, 33004–33015. [Google Scholar] [CrossRef] [Green Version]
- Zahreddine, H.A.; Culjkovic-Kraljacic, B.; Assouline, S.; Gendron, P.; Romeo, A.A.; Morris, S.J.; Cormack, G.; Jaquith, J.B.; Cerchietti, L.; Cocolakis, E.; et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014, 511, 90–93. [Google Scholar] [CrossRef] [Green Version]
- Norsworthy, K.J.; By, K.; Subramaniam, S.; Zhuang, L.; Del Valle, P.L.; Przepiorka, D.; Shen, Y.L.; Sheth, C.M.; Liu, C.; Leong, R.; et al. FDA Approval Summary: Glasdegib for Newly Diagnosed Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 6021–6025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becher, O.J.; Hambardzumyan, D.; Fomchenko, E.I.; Momota, H.; Mainwaring, L.; Bleau, A.-M.; Katz, A.M.; Edgar, M.; Kenney, A.M.; Cordon-Cardo, C.; et al. Gli Activity Correlates with Tumor Grade in Platelet-Derived Growth Factor–Induced Gliomas. Cancer Res. 2008, 68, 2241–2249. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Xie, G.; Fan, Q.; Xie, J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 2010, 29, 469–481. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Xin, L.; Liang, A.; Fu, Y. Cancer stem cell hypothesis: A brief summary and two proposals. Cytotechnology 2013, 65, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Shigdar, S.; Gantier, M.P.; Hou, Y.; Wang, L.; Li, Y.; Shamaileh, H.A.; Yin, W.; Zhou, S.F.; Zhao, X.; et al. Cancer stem cell targeted therapy: Progress amid controversies. Oncotarget 2015, 6, 44191–44206. [Google Scholar] [CrossRef] [Green Version]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Butti, R.; Gunasekaran, V.P.; Kumar, T.V.S.; Banerjee, P.; Kundu, G.C. Breast cancer stem cells: Biology and therapeutic implications. Int. J. Biochem. Cell Biol. 2019, 107, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Gulino, A.; Ferretti, E.; De Smaele, E. Hedgehog signalling in colon cancer and stem cells. EMBO Mol. Med. 2009, 1, 300–302. [Google Scholar] [CrossRef]
- Po, A.; Silvano, M.; Miele, E.; Capalbo, C.; Eramo, A.; Salvati, V.; Todaro, M.; Besharat, Z.M.; Catanzaro, G.; Cucchi, D.; et al. Noncanonical GLI1 signaling promotes stemness features and in vivo growth in lung adenocarcinoma. Oncogene 2017, 36, 4641–4652. [Google Scholar] [CrossRef] [Green Version]
- Sneha, S.; Nagare, R.P.; Sidhanth, C.; Krishnapriya, S.; Garg, M.; Ramachandran, B.; Murhekar, K.; Sundersingh, S.; Ganesan, T.S. The hedgehog pathway regulates cancer stem cells in serous adenocarcinoma of the ovary. Cell Oncol. 2020, 43, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Lytle, N.K.; Ferguson, L.P.; Rajbhandari, N.; Gilroy, K.; Fox, R.G.; Deshpande, A.; Schürch, C.M.; Hamilton, M.; Robertson, N.; Lin, W.; et al. A Multiscale Map of the Stem Cell State in Pancreatic Adenocarcinoma. Cell 2019, 177, 572–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.H.; Chen, B.Y.; Wu, C.Y.; Tsao, Z.J.; Chen, Y.Y.; Chang, C.P.; Yang, C.R.; Lin, D.P. Hedgehog overexpression leads to the formation of prostate cancer stem cells with metastatic property irrespective of androgen receptor expression in the mouse model. J. Biomed. Sci. 2011, 18, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikegame, A.; Ozaki, S.; Tsuji, D.; Harada, T.; Fujii, S.; Nakamura, S.; Miki, H.; Nakano, A.; Kagawa, K.; Takeuchi, K.; et al. Small molecule antibody targeting HLA class I inhibits myeloma cancer stem cells by repressing pluripotency-associated transcription factors. Leukemia 2012, 26, 2124–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Peng, C.; Sullivan, C.; Li, D.; Li, S. Critical molecular pathways in cancer stem cells of chronic myeloid leukemia. Leukemia 2010, 24, 1545–1554. [Google Scholar] [CrossRef] [Green Version]
- Mar, B.G.; Amakye, D.; Aifantis, I.; Buonamici, S. The controversial role of the Hedgehog pathway in normal and malignant hematopoiesis. Leukemia 2011, 25, 1665–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robarge, K.D.; Brunton, S.A.; Castanedo, G.M.; Cui, Y.; Dina, M.S.; Goldsmith, R.; Gould, S.E.; Guichert, O.; Gunzner, J.L.; Halladay, J.; et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Wu, X.; Jiang, J.; Gao, W.; Wan, Y.; Cheng, D.; Han, D.; Liu, J.; Englund, N.P.; Wang, Y.; et al. Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist. ACS Med. Chem. Lett. 2010, 1, 130–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlou, M.; Lu, J.F.; Wu, G.; Maity, S.; Tzelepi, V.; Navone, N.M.; Hoang, A.; Logothetis, C.J.; Efstathiou, E. Hedgehog signaling inhibition by the small molecule smoothened inhibitor GDC-0449 in the bone forming prostate cancer xenograft MDA PCa 118b. Prostate 2012, 72, 1638–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katagiri, S.; Tauchi, T.; Okabe, S.; Minami, Y.; Kimura, S.; Maekawa, T.; Naoe, T.; Ohyashiki, K. Combination of ponatinib with Hedgehog antagonist vismodegib for therapy-resistant BCR-ABL1-positive leukemia. Clin. Cancer Res. 2013, 19, 1422–1432. [Google Scholar] [CrossRef] [Green Version]
- Mimeault, M.; Rachagani, S.; Muniyan, S.; Seshacharyulu, P.; Johansson, S.L.; Datta, K.; Lin, M.F.; Batra, S.K. Inhibition of hedgehog signaling improves the anti-carcinogenic effects of docetaxel in prostate cancer. Oncotarget 2015, 6, 3887–3903. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: Molecular mechanisms. PLoS ONE 2011, 6, e27306. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Mysliwietz, J.; Ellwart, J.; Gamarra, F.; Huber, R.M.; Bergner, A. Effects of the Hedgehog pathway inhibitor GDC-0449 on lung cancer cell lines are mediated by side populations. Clin. Exp. Med. 2012, 12, 25–30. [Google Scholar] [CrossRef]
- Wang, C.; Wu, H.; Katritch, V.; Han, G.W.; Huang, X.P.; Liu, W.; Siu, F.Y.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Structure of the human smoothened receptor bound to an antitumour agent. Nature 2013, 497, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.; Alicke, B.; West, K.A.; Pacheco, P.; La, H.; Januario, T.; Yauch, R.L.; de Sauvage, F.J.; Gould, S.E. Pharmacokinetic-pharmacodynamic analysis of vismodegib in preclinical models of mutational and ligand-dependent Hedgehog pathway activation. Clin. Cancer Res. 2011, 17, 4682–4692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumthekar, P.U.; Macrie, B.D.; Singh, S.K.; Kaur, G.; Chandler, J.P.; Sejpal, S.V. A review of management strategies of malignant gliomas in the elderly population. Am. J. Cancer Res. 2014, 4, 436–444. [Google Scholar] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro-Oncol. 2013, 15 (Suppl. 2), ii1–ii56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopschinski, B.E.; Beier, C.P.; Beier, D. Glioblastoma cancer stem cells--from concept to clinical application. Cancer Lett. 2013, 338, 32–40. [Google Scholar] [CrossRef]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Twigg, S.R.F.; Hufnagel, R.B.; Miller, K.A.; Zhou, Y.; McGowan, S.J.; Taylor, J.; Craft, J.; Taylor, J.C.; Santoro, S.L.; Huang, T.; et al. A Recurrent Mosaic Mutation in SMO, Encoding the Hedgehog Signal Transducer Smoothened, Is the Major Cause of Curry-Jones Syndrome. Am. J. Hum. Genet. 2016, 98, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- Musani, V.; Gorry, P.; Basta-Juzbasic, A.; Stipic, T.; Miklic, P.; Levanat, S. Mutation in exon 7 of PTCH deregulates SHH/PTCH/SMO signaling: Possible linkage to WNT. Int. J. Mol. Med. 2006, 17, 755–759. [Google Scholar] [CrossRef] [Green Version]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.S.; et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoni, M.; Burattini, L.; Nabissi, M.; Morelli, M.B.; Berardi, R.; Santoni, G.; Cascinu, S. Essential role of Gli proteins in glioblastoma multiforme. Curr. Protein Pept. Sci. 2013, 14, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, K.; Huang, L.; Liu, C.; Du, Q.; Li, T.; Yan, C.; Feng, Z.; Li, X. Anticancer effects of FL34 through the inhibition of GLI1 in glioblastoma. J. Cancer Res. Ther. 2019, 15, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, Y.; Gao, R.; Yu, H.; Sun, T. HDAC6 inhibition induces glioma stem cells differentiation and enhances cellular radiation sensitivity through the SHH/Gli1 signaling pathway. Cancer Lett. 2018, 415, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Cremers, N.; Kroese, F.; Orend, G.; Chiquet-Ehrismann, R.; Uede, T.; Yagita, H.; Sleeman, J.P. CD24 expression causes the acquisition of multiple cellular properties associated with tumor growth and metastasis. Cancer Res. 2005, 65, 10783–10793. [Google Scholar] [CrossRef] [Green Version]
- Runz, S.; Mierke, C.T.; Joumaa, S.; Behrens, J.; Fabry, B.; Altevogt, P. CD24 induces localization of beta1 integrin to lipid raft domains. Biochem. Biophys. Res. Commun. 2008, 365, 35–41. [Google Scholar] [CrossRef]
- Karaman, S.; Leppänen, V.M.; Alitalo, K. Vascular endothelial growth factor signaling in development and disease. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Wu, Q.; Li, J.; Zhu, S.; Wu, J.; Chen, C.; Liu, Q.; Wei, W.; Zhang, Y.; Sun, S. Breast cancer subtypes predict the preferential site of distant metastases: A SEER based study. Oncotarget 2017, 8, 27990–27996. [Google Scholar] [CrossRef] [Green Version]
- Hatsell, S.; Frost, A.R. Hedgehog signaling in mammary gland development and breast cancer. J. Mammary Gland Biol. Neoplasia 2007, 12, 163–173. [Google Scholar] [CrossRef]
- Hatsell, S.J.; Cowin, P. Gli3-mediated repression of Hedgehog targets is required for normal mammary development. Development 2006, 133, 3661–3670. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, S.A.; Machalek, D.A.; Shearer, R.F.; Millar, E.K.; Nair, R.; Schofield, P.; McLeod, D.; Cooper, C.L.; McNeil, C.M.; McFarland, A.; et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res. 2011, 71, 4002–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkkonen, T.; Lewis, M.T. New paradigms for the Hedgehog signaling network in mammary gland development and breast Cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 315–332. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Johnson, R.L.; Zhang, X.; Bare, J.W.; Waldman, F.M.; Cogen, P.H.; Menon, A.G.; Warren, R.S.; Chen, L.C.; Scott, M.P.; et al. Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer Res. 1997, 57, 2369–2372. [Google Scholar]
- Jiao, X.; Wood, L.D.; Lindman, M.; Jones, S.; Buckhaults, P.; Polyak, K.; Sukumar, S.; Carter, H.; Kim, D.; Karchin, R.; et al. Somatic mutations in the Notch, NF-KB, PIK3CA, and Hedgehog pathways in human breast cancers. Genes Chromosomes Cancer 2012, 51, 480–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, S.K.; Khan, J.S.; Shah, S.T.A.; Wang, F.; Ye, L.; Jiang, W.G.; Malik, M.F.A. Involvement of hedgehog pathway in early onset, aggressive molecular subtypes and metastatic potential of breast cancer. Cell Commun. Signal. 2018, 16, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noman, A.S.; Uddin, M.; Rahman, M.Z.; Nayeem, M.J.; Alam, S.S.; Khatun, Z.; Wahiduzzaman, M.; Sultana, A.; Rahman, M.L.; Ali, M.Y.; et al. Overexpression of sonic hedgehog in the triple negative breast cancer: Clinicopathological characteristics of high burden breast cancer patients from Bangladesh. Sci. Rep. 2016, 6, 18830. [Google Scholar] [CrossRef]
- Koike, Y.; Ohta, Y.; Saitoh, W.; Yamashita, T.; Kanomata, N.; Moriya, T.; Kurebayashi, J. Anti-cell growth and anti-cancer stem cell activities of the non-canonical hedgehog inhibitor GANT61 in triple-negative breast cancer cells. Breast Cancer 2017, 24, 683–693. [Google Scholar] [CrossRef]
- Xu, L.; Kwon, Y.J.; Frolova, N.; Steg, A.D.; Yuan, K.; Johnson, M.R.; Grizzle, W.E.; Desmond, R.A.; Frost, A.R. Gli1 promotes cell survival and is predictive of a poor outcome in ERalpha-negative breast cancer. Breast Cancer Res. Treat. 2010, 123, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sanchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Balko, J.M.; Schwarz, L.J.; Luo, N.; Estrada, M.V.; Giltnane, J.M.; Davila-Gonzalez, D.; Wang, K.; Sanchez, V.; Dean, P.T.; Combs, S.E.; et al. Triple-negative breast cancers with amplification of JAK2 at the 9p24 locus demonstrate JAK2-specific dependence. Sci. Transl. Med. 2016, 8, 334ra53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar] [CrossRef] [Green Version]
- Lorger, M.; Felding-Habermann, B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am. J. Pathol. 2010, 176, 2958–2971. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Pisano, C.; Vlodavsky, I.; Ilan, N.; Zunino, F. The potential of heparanase as a therapeutic target in cancer. Biochem. Pharmacol. 2014, 89, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Vlodavsky, I.; Ilan, N.; Sanderson, R.D. Forty Years of Basic and Translational Heparanase Research. Adv. Exp. Med. Biol. 2020, 1221, 3–59. [Google Scholar] [CrossRef] [Green Version]
- Lanzi, C.; Zaffaroni, N.; Cassinelli, G. Targeting Heparan Sulfate Proteoglycans and their Modifying Enzymes to Enhance Anticancer Chemotherapy Efficacy and Overcome Drug Resistance. Curr. Med. Chem. 2017, 24, 2860–2886. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Chatterjee, M.; Grasso, M.; Specchia, G.; Magen, H.; Einsele, H.; Celeghini, I.; Barbieri, P.; Paoletti, D.; Pace, S.; et al. Phase I study of the heparanase inhibitor roneparstat: An innovative approach for ultiple myeloma therapy. Haematologica 2018, 103, e469–e472. [Google Scholar] [CrossRef]
- Ramani, V.C.; Zhan, F.; He, J.; Barbieri, P.; Noseda, A.; Tricot, G.; Sanderson, R.D. Targeting heparanase overcomes chemoresistance and diminishes relapse in myeloma. Oncotarget 2016, 7, 1598–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saman, H.; Raza, S.S.; Uddin, S.; Rasul, K. Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches. Cancers 2020, 12, 1172. [Google Scholar] [CrossRef]
- Gordon, M.S.; Margolin, K.; Talpaz, M.; Sledge, G.W., Jr.; Holmgren, E.; Benjamin, R.; Stalter, S.; Shak, S.; Adelman, D. Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J. Clin. Oncol. 2001, 19, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Zirlik, K.; Duyster, J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol. Res. Treat. 2018, 41, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Scartozzi, M.; Vincent, L.; Chiron, M.; Cascinu, S. Aflibercept, a New Way to Target Angiogenesis in the Second Line Treatment of Metastatic Colorectal Cancer (mCRC). Target. Oncol. 2016, 11, 489–500. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Isoform | Target Gene | Cancer | Reference |
|---|---|---|---|
| GLI1 | ABCG2 | Diffuse large B-cell lymphoma | [140] |
| ANO1 | Pancreatic ductal adenocarcinoma | [141] | |
| AQP1 | Glioma | [142] | |
| Bcl-2 | Colorectal adenocarcinoma, basal cell carcinoma, B-cell chronic lymphocytic leukemia | [143,144,145] | |
| BHLHE41 | Pancreatic ductal adenocarcinoma | [146] | |
| Bmi1 | Medulloblastoma | [147] | |
| c-Myc | Pancreatic adenocarcinoma | [148] | |
| CXCR4 | Breast cancer, pancreatic ductal adenocarcinoma | [149,150] | |
| Cyclin D1 | Pancreatic ductal adenocarcinoma | [151] | |
| Cyclin D2 | Rhabdomyosarcoma, medulloblastoma, astrocytoma, cervical cancer | [152,153,154] | |
| DMNT1 | Pancreatic ductal adenocarcinoma | [155] | |
| ENC1 | Medulloblastoma, rhabdomyosarcoma | [156] | |
| E2F1 | Melanoma | [157] | |
| FOXM1 | Basal cell carcinoma, bladder cancer, glioblastoma | [158,159,160] | |
| FOXS1 | Medulloblastoma, rhabdomyosarcoma | [156] | |
| GLI1 | B-cell chronic lymphocytic leukemia, chronic myelogeneous leukemia, medulloblastoma, multiple myeloma, rhabdomyosarcoma | [145,156,161,162] | |
| GLI2 | B-cell chronic lymphocytic leukemia, multiple myeloma | [145,162] | |
| H19 | Bladder cancer | [158] | |
| HHIP | Medulloblastoma, rhabdomyosarcoma | [163] | |
| IGF2 | Bladder cancer | [158] | |
| IGFBP-6 | Rhabdomyosarcoma, neuroblastoma, colon cancer | [164] | |
| IL-7 | Pancreatic ductal carcinoma | [151] | |
| KLF4 | Colon cancer | [165] | |
| Krox-20 | Medulloblastoma, cervical cancer | [153] | |
| MUC5AC | Pancreatic ductal adenocarcinoma | [166] | |
| Nanog | Colon cancer, glioblastoma, medulloblastoma, pancreatic adenocarcinoma | [148,165,167,168] | |
| NEO1 | Basal cell carcinoma, medulloblastoma | [169,170] | |
| NKX2.2 | Medulloblastoma, astrocytoma | [154] | |
| OCT4 | Colon cancer, pancreatic adenocarcinoma | [148,165] | |
| Osteopontin | Melanoma | [164] | |
| PAX6 | Medulloblastoma, astrocytoma | [154] | |
| PDGFRα | Basal cell carcinoma | [171] | |
| Plakoglobin | Rhabdomyosarcoma, medulloblastoma, astrocytoma | [152,154] | |
| PLAT | Medulloblastoma, rhabdomyosarcoma | [156] | |
| PRPSAP1 | Cervical cancer | [153] | |
| PSF2 | Bladder cancer | [158] | |
| PTCH1 | Chronic myelogeneous leukemia, medulloblastoma, multiple myeloma, rhabdomyosarcoma | [154,156,161,162] | |
| PTCH2 | Medulloblastoma, rhabdomyosarcoma | [156] | |
| SHH | Chronic myelogeneous leukemia | [161] | |
| SMO | Chronic myelogeneous leukemia | [161] | |
| Snail1 | Melanoma | [172] | |
| SOSTDC1 | Medulloblastoma, rhabdomyosarcoma | [156] | |
| Sox2 | Colon cancer, non-small-cell lung cancer, pancreatic ductal adenocarcinoma | [148,165,173] | |
| SPP1 | Bladder, melanoma | [158] | |
| Twist1 | Melanoma | [172] | |
| WNT-2 | Colon cancer | [165] | |
| Zeb1 | Melanoma | [172] | |
| tGLI1 | CD24 | Breast, glioblastoma | [24,139] |
| CD44 | Breast, glioblastoma | [31] | |
| Cep70 1 | Breast cancer | [32] | |
| HPSE | Glioblastoma | [29] | |
| MMP-2 | Breast cancer | [139] | |
| MMP-9 | Breast cancer | [139] | |
| Nanog | Breast cancer | [33] | |
| OCT4 | Breast cancer | [33] | |
| PTCH1 | Glioblastoma | [24] | |
| R-Ras2 1 | Breast cancer | [32] | |
| Sox2 | Breast cancer | [33] | |
| TEM7 | Glioblastoma | [30] | |
| UPF3A 1 | Breast cancer | [32] | |
| VEGF-A | Breast, glioblastoma | [139,174] | |
| VEGF-C | Glioblastoma | [30] | |
| VEGFR2 | Breast, medulloblastoma | [28] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doheny, D.; Manore, S.G.; Wong, G.L.; Lo, H.-W. Hedgehog Signaling and Truncated GLI1 in Cancer. Cells 2020, 9, 2114. https://doi.org/10.3390/cells9092114
Doheny D, Manore SG, Wong GL, Lo H-W. Hedgehog Signaling and Truncated GLI1 in Cancer. Cells. 2020; 9(9):2114. https://doi.org/10.3390/cells9092114
Chicago/Turabian StyleDoheny, Daniel, Sara G. Manore, Grace L. Wong, and Hui-Wen Lo. 2020. "Hedgehog Signaling and Truncated GLI1 in Cancer" Cells 9, no. 9: 2114. https://doi.org/10.3390/cells9092114