Podoplanin as an Attractive Target of CAR T Cell Therapy


Shin Kaneko Laboratory, Department of Cell Growth and Differentiation, Center for iPS Cell Research and Application (CiRA), Kyoto University, Kyoto 606-8507, Japan
*
Author to whom correspondence should be addressed.
Cells 2020, 9(9), 1971; https://doi.org/10.3390/cells9091971
Submission received: 15 July 2020
/
Revised: 24 August 2020
/
Accepted: 24 August 2020
/
Published: 26 August 2020
(This article belongs to the Special Issue Structure and Function of Podoplanin (PDPN) in Disease)
{kind=link}
{kind=link}
Abstract
:To date, various kinds of cancer immunotherapy methods have been developed, but T cell immunotherapy is one of the most promising strategies. In general, T cell receptor (TCR) or chimeric antigen receptor (CAR) is used to modify the antigen specificity of T cells. CARs possess an underlying potential with treatment efficacy to treat a broad range of cancer patients compared with TCRs. Although a variety of CAR molecules have been developed so far, the clinical application for solid tumors is limited partly due to its adverse effect known as “on-target off-tumor toxicity”. Therefore, it is very important for CAR T cell therapy to target specific antigens exclusively expressed by malignant cells. Here, we review the application of T cell immunotherapy using specific antigen receptor molecules and discuss the possibility of the clinical application of podoplanin-targeted CAR derived from a cancer-specific monoclonal antibody (CasMab).
1. Introduction
In our body, there exist numerous varieties of immune cells, such as T cells, B cells, natural killer (NK) cells, granulocytes, macrophages, and dendritic cells, and they all act to protect us from different kinds of infections and diseases. Utilizing methods to enhance or to activate these immune cells in order to treat cancer is a fundamental strategy that is extensively applied in cancer immunotherapy. To date, various kinds of cancer immunotherapy methods have been developed, but one method that has been extensively applied in research and development is the use of T cells.
T cells are derived from hematopoietic stem cells that differentiate in the thymus. Once migrated to the thymus, T-cell progenitors proliferate in response to interleukin-7 (IL-7) and Delta-like 4 (DLL4), and begin to differentiate into T cells during this process [1,2]. Inside the T-cell progenitors that are stimulated by IL-7 and DLL4, the T cell receptor (TCR) gene locus undergoes rearrangement forming a diverse repertoire; however, appropriate selection within the thymus results in the formation of T cells expressing specific TCRs that are responsible for appropriate responsiveness after being chosen, and then they migrate into the periphery.
Cytotoxic T lymphocytes (CTLs) are the specific T cells that express CD8, and via the TCR they can distinguish between the complexes of antigen peptides and human leukocyte antigens (HLAs) that are present on the cell surface, and thereby possess the function of attacking abnormal cells in an antigen-specific manner. Consequently, it is believed that generating large numbers of CTLs that can specifically recognize cancer cells would enable us to develop effective treatments for specifically targeting the cancer cells. In this review article, we will outline the application of cancer immunotherapy using CTLs with incorporated antigen recognition receptors and explain the possibility of the clinical application of chimeric antigen receptor (CAR) T cell therapies that target the mucin-type glycoprotein, podoplanin (PDPN), and finally the possibility of using CAR T cell therapies that would leverage the new technology of induced pluripotent stem (iPS) cells.
2. Antigen Receptor-Transduced CTL Therapy
Tumor infiltrating lymphocytes (TILs) that are specifically responsive to cancer cells were shown to exist in the tumors that were excised from the patients, and since then, several reports were released regarding the possibility to expand TILs ex vivo and use them in cancer treatment [3]. Thereafter, research on TILs has drawn the attention of many researchers. In recent research, it has clearly been shown that the state of differentiation of T cells used for the treatment and their effectiveness for cancer treatment are closely related, thereby increasing the importance of methods that involve selective proliferation of T cells at an early stage of differentiation and introducing genes into them [4]. However, the methods and duration of treatments for each patient differ, and it might be because a sufficient number of T cells cannot be obtained in an early state of differentiation from the patient’s TILs. Consequently, an alternate strategy was devised whereby TCR genes were cloned from cancer cell-specific TILs and transduced into the less or moderately differentiated T cells for the purpose of treatment, and this has been clinically tested.
In many cancer cells, various gene mutations have been shown to accumulate in the genome. Some of these genetic mutations are responsible for expressing specific antigens called “neoantigens”. Interestingly, it has been found that TILs include certain T cells that can specifically recognize these neoantigens [5]. Methods have been developed for determining the neoantigen-specific TCRs by combining various screening systems with sequencing technologies including whole exome sequencing and gene expression analysis of the normal tissue and tumor tissue derived from the same patients [6]. With the advancements in the next-generation sequencing technologies and improvements in the accuracy of antigen prediction algorithms, it is becoming possible to rapidly generate CTLs that are equipped with neoantigen-specific TCRs and can target the specific tumor cells of individual patients.
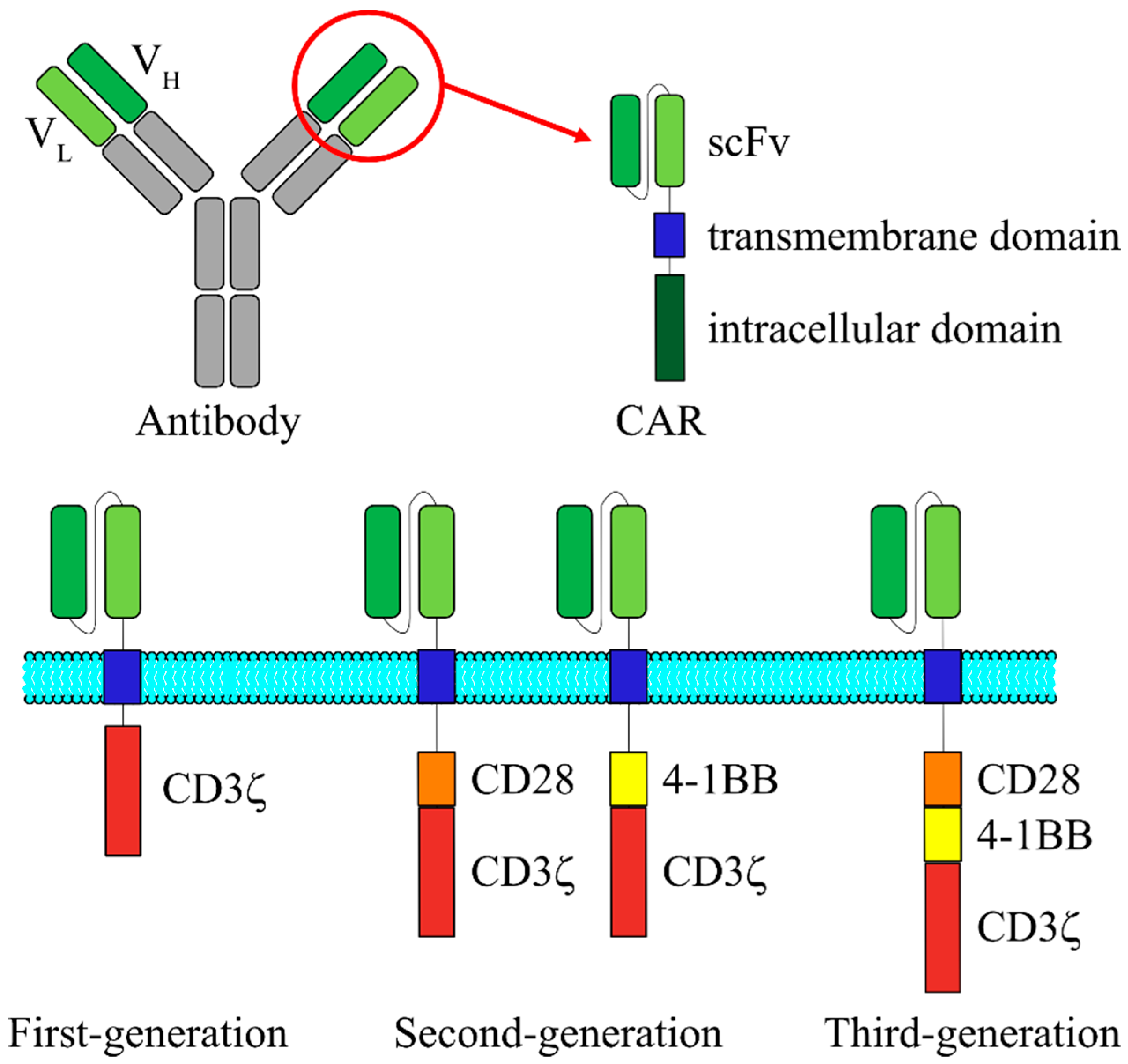
Meanwhile, a new antigen recognition receptor called “CAR” has been developed, and this novel receptor is being extensively used for the clinical applications. In general, CAR-expressing T cells are prepared by the following procedures: the desired T cells are harvested and purified from the apheresis product from the patient or an allogeneic donor, followed by T cell activation on anti-CD3/28 beads serving as artificial antigen-presenting cells. The activated T cells are then genetically modified ex vivo by introduction of a gene encoding CAR molecule, and the CAR transduced cells are further expanded ex vivo. When the genetically modified T-cell product has been prepared, the patient receives lymphodepleting preparative regimen followed by adoptive transfer of CAR T cells. CAR is an artificial protein with a basic structure that is comprised of a single chain variable fragment (scFv) derived from an antibody on the N-terminal and a CD3ζ chain on the C-terminal (Figure 1). Between these two terminals, a hinge and a transmembrane domain are also incorporated [7]. First-generation CARs, which do not possess costimulatory receptor-derived signal transduction domain, exhibit high cytotoxic efficacy against target cells in vitro, but exhibit almost no cytotoxic efficacy in clinical trials. This was believed to be due to the poor potential of T cell proliferation after activation, which would further prevent the CAR T cells from acting for longer durations within the patients [8,9]. In contrast, in addition to the composition of first-generation CARs, second-generation CARs are shown to possess one of the costimulatory receptor-derived signal transduction domains, such as from CD28 and 4-1BB, and the CAR T cells did not lose the proliferation capacity even when they were repeatedly stimulated. Therefore, they demonstrated excellent IL-2 release and cell proliferation characteristics after the stimulation [10,11,12]. Third-generation CARs possess two costimulatory receptor-derived signal transduction domains and have been reported to succeed in cytokine release following antigen stimulation [13]. In many different reports using second- and third-generation CARs, it has not been clearly demonstrated: which one is better for CAR T therapy? However, compared to the first-generation CARs, T cells with CARs of the second and third generations have been shown to exhibit better proliferation, cytokine production, and cytotoxic activity, and longer survival in vivo [14,15,16].
Since TCR can target the complexes of HLA and the peptides that are presented, treatment is possible by using only the TCR when the patients possess specific HLAs (HLA restriction). However, it is common to observe a decline or elimination of HLA expression on cancer cells, which is a well-known mechanism whereby cancer cells escape from the immune system [17]. Consequently, CTL therapies using TCR are required to overcome the issue of HLA expression. In contrast, since CARs do not have HLA restriction, they have the advantage of being used for the treatment of any kind of cancer patients as long as the cancer cells can express the target antigen. In addition, for the activation of T cells, it is necessary to have co-signaling via costimulatory receptors in addition to the signaling induced by TCR. Moreover, by using the CARs of the second-generation or later, it is possible to generate both the signals simultaneously with a single CAR, so that they exhibit cytotoxicity against cancer cells that do not express ligands for the costimulatory receptors.
More recent studies have suggested the potential application of CAR T cell therapy for other diseases. For example, Aghajanian et al. reported the efficacy of CAR T cell therapy for this disease using a mouse model of myocardial fibrosis [18]. Fibrosis is a condition that is observed in conjunction with numerous myocardial diseases, and activated fibroblasts produce excessive extracellular matrix proteins. However, while there is now a therapeutic method that directly targets fibrosis, it has limitations, and new treatment methods are needed. Therefore, to discover new therapeutic targets, gene expression analysis was performed in cardiac tissues obtained from heart transplant donors and recipients using an RNA sequence analysis database; fibroblast activation protein (FAP) was identified as a target candidate as a result. FAP is a glycoprotein that is expressed in fibrotic tissues and various cancer cells but is rarely expressed in normal human and mouse tissues. FAP has also been discussed in reports from studies where it is used as a target for cancer treatments using CAR T cells [19,20,21]. As a result of human and murine cardiac tissue staining, FAP was found to be strongly expressed in fibroblasts present in myocardial fibrotic tissue. Therefore, when CAR T cells targeting FAP were transplanted into myocardial fibrosis model mice, fibrosis was significantly suppressed as compared with the non-treated group, and the cardiac function was equivalent to that of normal tissue.
In this way, CARs possess an underlying potential with treatment efficacy to treat a broad range of patients not only with cancer but also with other diseases compared to TCRs.
3. Extending CAR T Cell Therapy from B Cell Malignancies to Solid Tumors
The clinical trials that have currently progressed extensively are performed for the CAR T cell therapies that target hematologic malignancies with B cells that express CD19. CD19 is expressed in tumor cells, such as in non-Hodgkin’s lymphoma, acute lymphoblastic leukemia (ALL), and chronic lymphocytic leukemia (CLL), but is not expressed in hematopoietic stem cells, non-B-cell hematopoietic cells, and non-lymphoid tissues. Previously, a high rate of complete remission has been obtained in phase I and II clinical trials, targeting the children or young patients with relapsing and intractable CD19-positive ALL. Additionally, there have been reports of cases where remission was maintained for long periods, such as over 2 years since the treatment [22]. Confirming its considerable clinical efficacy, treatment with CAR T cells that target this molecule was approved by the U.S. Food and Drug Administration (FDA) in 2017, and the development of CAR T cell treatments that target hematological malignancies is being pursued actively worldwide.
However, previous studies have suggested that there are at least two patterns in which CAR T cell therapies are ineffective [22]. The first involves instances where transplanted CAR T cells do not actively proliferate and thus are unable to exert a sufficient therapeutic effect. For example, it is believed that T cells, which are the source of CAR T cells, can become exhausted by treatments patients previously received, such as radiotherapies and anticancer chemotherapy. Therefore, in TCR-introduced T cell therapy, it is important to select T cells in an appropriate state to generate CAR T cells. The second is when cancer cells evade CAR T cell attack by mutation or loss of expression of the CD19 antigen epitope of CAR T cells [23]. For example, in a Phase II clinical trial investigating pediatric and adolescent patients with relapsed or refractory B-cell acute lymphoblastic leukemia (B-ALL), only one patient bearing CD19-positive leukemia cells relapsed, while fifteen cases of CD19-negative recurrence were confirmed, which is an important issue in achieving complete remission of the disease [24]. Studies of CAR T cell therapies targeting multiple antigens are currently ongoing as part of a strategy to overcome this issue. To date, 4-1BB-based second-generation CAR T cells targeting antigens such as CD20, CD22, CD123, in addition to CD19, have been evaluated using a xenograft model. In these studies, CAR T cells targeting two different antigens have been shown to significantly suppress recurrence due to CD19-negative cells in comparison to CAR T cells targeting CD19 alone [25,26,27]. Further, it has been reported as a result of clinical studies that the administration of CAR T cells targeting CD19 and CD22 can improve the therapeutic effect [28]. In addition, by combining fluorescein isothiocyanate (FITC)-labeled antibodies and anti-FITC CAR proteins, a method for addressing multiple target molecules using a single CAR has been developed, which enables simplified targeting of multiple antigens [29]. As such, CAR T cell therapies targeting multiple antigens are expected to become mainstream in the future to prevent the recurrence of tumor cells that have lost antigenicity.
However, in contrast to the clear clinical efficacy of CAR T cells against the hematological malignancies, the clinical efficacy of CAR T cell therapies against solid tumors has not been established yet. Major factors contributing to this are the need to overcome the mechanisms of immunosuppression in the tumor microenvironment and the requirement of accumulating the CAR T cells at the tumor site. To date, immunotherapy involving IL-2 administration has been widely used to enhance the antitumor effect, and a certain level of efficacy has been observed in metastatic melanoma and renal cell carcinoma patients [30]. However, previous studies have shown that IL-2 is involved in activation-induced cell death, suppression of memory T cell proliferation and survival, and the proliferation of regulatory T cells, and it has also been shown that there is a risk of death associated with further treatment related to high-concentration IL-2 administration [31,32,33,34]. As such, a new T cell activator capable of avoiding these issues is required, and IL-15 is currently considered to be one of the most promising candidates. Similar to IL-2, IL-15 uses a complex containing CD122 and CD132 as a receptor and it is known to show similar properties than IL-2, such as the ability to induce T cell proliferation. Meanwhile, various types of IL-15, such as secretory IL-15, secretory IL-15/IL-15R complex, and membrane-bound IL-15, have been studied. IL-15 has been reported to maintain memory T cells and suppress apoptosis [35,36,37]. Furthermore, co-expression of IL-15 in CAR T cells has been confirmed to markedly improve the antitumor effect in xenograft model mice [38,39]. In addition, there was also a report that the generation of CAR T cells that simultaneously express IL-7, which is known to promote T cell proliferation and survival, and CCL19, which is a chemoattractant of T cells and dendritic cells, was successful in obtaining an extremely high antitumor effect [40]. As described above, studies using various humoral factors other than IL-2 are ongoing worldwide, and there have been numerous attempts to find practical applications for CAR T cells capable of remarkable antitumor effects against solid tumors. An additional issue is that solid tumors are highly heterogeneous, which makes it difficult to identify a suitable, single target molecule [41].
4. Adverse Effects and Toxicities of CAR T Cell Therapy
The most prominent toxicities of CAR T cells are cytokine release syndrome (CRS) and central nervous system (CNS) complications. CRS is thought to be caused by cytokines secreted by administered T cells, and a series of symptoms including fever of over 39 °C and hypotension were observed mainly from several days to 1–2 weeks after administration [42,43]. However, neurological toxicities may occur at a different time or independently of CRS, and it is speculated that this may be due to a mechanism different from CRS in at least some cases. As mild to moderate neurological side effects, various symptoms such as headache, mild aphasia, movement disorder, encephalopathy, and delirium have been reported, but in severe cases, the occurrence of serious symptoms such as coma, intracranial hemorrhage, and brain edema was reported [44,45,46].
Although many mechanisms have not yet been fully characterized, our pathophysiological understanding of CRS and CNS complications continues to grow. For example, recent findings from a study of the murine xenograft model suggest that not only CAR T cells but also monocytes/macrophages play important roles in the development of CRS and neurological side effects [47]. Specifically, in humanized mice with a high leukemia burden, the main source of IL-1 and IL-6 during CRS development was human-derived monocytes. Although the inhibition of IL-6 receptors with tocilizumab prevented the onset of CRS, CNS complications were still confirmed. By contrast, the inhibition of IL-1 signal transduction using anakinra, an IL-1 receptor antagonist, was able to suppress both CRS and CNS complications. As IL-1 secretion precedes IL-6 production, and IL-1 induces IL-6 secretion, IL-1 is believed to facilitate the release of IL-6 into the bloodstream in the patients with CRS. The excessive release of cytokines into the bloodstream by the activation of CAR T cells and monocytes/macrophages stimulates endothelial cells exposed to high concentrations of IL-6 and IFN-γ [48,49]. When vascular endothelial cells throughout the body are activated and vascular permeability is enhanced, blood pressure falls and hypoalbuminemia is induced, resulting in systemic capillary leakage syndrome. In vitro experiments have also shown that IL-6 disrupts the blood–brain barrier (BBB)—by acting on vascular endothelial cells and promoting a decrease in the expression of tight junction-related molecules [50]. In vitro findings at the BBB are supported by results from studies of patients who developed fatal cerebral edema and had severe CRS and CNS complications [44,51]. Based on these findings, the development of novel therapeutic agents that are able to suppress the enhancement of vascular permeability and BBB breakdown, as well as prevent CRS and CNS complications, can be expected to make further advancements.
Although no optimal treatment for CRS has been established, previous studies have shown that anti-IL-6 receptor antibody therapy and corticosteroid administration show specific effects. CRS has been observed to be associated with the elevation of IL-6 in the early stage of clinical trials using CAR T cells, which has motivated researchers to try tocilizumab, an inhibitory antibody targeting the IL-6 receptor [52]. Currently, tocilizumab is the standard treatment for CRS and was approved by the FDA in 2017. So far, no adverse events have been reported with tocilizumab administration for CRS. There was also a concern that the antitumor activity of CAR T cells may be inhibited by the early administration of tocilizumab, but there is currently no evidence supporting this claim. The main limitation of tocilizumab is its inability to cross the BBB. Accordingly, CNS complications have been reported to still develop even after recovery from CRS by tocilizumab administration [24,53]. In fact, blood levels of IL-6 become elevated even after the IL-6 receptor is blocked by tocilizumab, and the brain will continue to be exposed to high levels of IL-6. Therefore, a method of directly inhibiting IL-6 by administering the anti-IL-6 antibody siltuximab to suppress CNS complications has also been investigated. Moreover, considering that it has a broad anti-inflammatory effect, corticosteroids are also an option in the treatment of CRS. However, given that corticosteroids are potentially T cell cytotoxic and this may affect their therapeutic efficacy, corticosteroids should be reserved for cases that are non-responsive to tocilizumab. Steroids are also frequently used to reduce CNS complications in cases that are non-responsive to tocilizumab monotherapy. Dexamethasone is also a strong candidate, considering its ability to reach the CNS.
Another serious issue with CAR T cell therapies is on-target off-tumor toxicity. This is because CAR T cells exhibit toxicity towards normal tissues expressing even small amounts of the target molecule. For example, there have been reports of respiratory distress and pulmonary infiltration, which were observed following the administration of ErbB2-specific CAR T cells, and consequently, the patients died 5 days after treatment. This is believed to be due to the low-level expression of ErbB2 on lung epithelial cells [54]. Furthermore, delayed-onset respiratory toxicity has been confirmed in clinical trials using CAR T cells targeting carcinoembryonic antigen related cell adhesion molecule 5 (CEACAM5), and on-target off-tumor toxicity has become a concern when tumor-associated antigens are targeted [55]. In another case, unexpected hepatotoxicity was also caused in a Phase I study targeting carbonic anhydrase IX (CAIX) expressed in renal cell carcinoma [56]. Although not seen in preclinical studies, normal bile duct epithelial cells are believed to express low levels of the CAIX antigen and are injured by CAR T cells. In addition, while clinical studies with CAR T cells targeting common antigens such as mesothelin, carcinoembryonic antigen (CEA), and ganglioside (GD2) have been conducted in the past, no remarkable toxicity was reported. However, virtually no antitumor activity was observed in these clinical studies, and it is highly probable that toxicity did not occur due to insufficient CAR T cell activity.
Various techniques have been developed to remove administered CAR T cells to address severe toxicity for instances unable to be controlled through the aforementioned treatment methods. One technique involves the expression of target antigenic molecules such as epidermal growth factor receptor (EGFR) and CD20 in CAR T cells and removal of CAR T cells administered using already clinically approved antibodies such as cetuximab and rituximab [57,58]. Another technique involves incorporating a “suicide gene” into CAR T cells. The system using inducible caspase-9 (iCasp9) produces a particularly rapid response, and its efficacy has been confirmed through clinical trials. iCasp9 is immediately dimerized by the administration of low molecular weight compounds, and it is possible to rapidly remove cells by activating the apoptotic pathway [59,60]. However, as these techniques have a substantial impact on antitumor efficacy, they are believed to be useful as a safety contingency in the case of lethal toxicity caused by CAR T cells that cannot be controlled by other methods. A recent report has stated that this activity can be regulated while maintaining CAR T cells in the body by using dasatinib, which inhibits multiple tyrosine kinases, but it will take time for a practical application to be developed [61]. Consequently, it is extremely important in CAR T cell therapies that only the antigens that are specifically expressed in cancer cells are applied.
5. PDPN and Anti-PDPN CasMab
Human podoplanin (PDPN) has a total length of 162 amino acids and consists of a highly glycosylated extracellular domain, a single-pass transmembrane domain, and a short intracellular domain of nine amino acids. PDPN itself is not known to have any enzymatic activity; however, it interacts with various other proteins, including C-type lectin-like receptor-2 (CLEC-2), CD44, galectin 8, C-C chemokine ligand 21 (CCL21), ezrin, moesin, protein kinase A (PKA), and cyclin dependent kinase 5 (CDK5), and has been reported to possess various other functions [62,63]. For example, PDPN colocalizes with the ezrin, radixin, and moesin (ERM) family proteins, and binds directly to ezrin and moesin. In addition, overexpression of PDPN leads to phosphorylation of ERM proteins and activation of RhoA, thereby increasing motility and induction of epithelial–mesenchymal transition (EMT) [64,65,66].
So far, research about PDPN has been carried out extensively, particularly in the field of cancer. PDPN acts as a molecular marker specific for lymphatic vessels and, because there is a correlation shown to exist between the formation of lymphatic vessels and poor cancer prognosis, it is also frequently used as a diagnostic marker for cancer [67,68]. In addition, increased expression of PDPN has been reported in numerous cases of solid tumors, including squamous cell carcinomas of the lungs, head and neck [69,70], malignant mesothelioma [71,72], and brain tumors [73]. PDPN is often identified to express at the leading edge of a tumor, suggesting that it contributes to metastasis and infiltration of the cancer cells [65]. Moreover, aggregates of circulating cancer cells with platelets through the binding of PDPN to CLEC-2 have been reported to further promote the process of hematogenous metastasis [74]. Therefore, inhibiting the interaction between PDPN and CLEC-2 is believed to be a unique, and feasible treatment strategy for targeting metastasis of the cancer cells [75].
NZ-1 is an anti-PDPN antibody and, in the research established by Kato et al., the authors used the antibodies derived from NZ-1 and demonstrated an anti-tumor effect using the xenograft models of glioma, mesothelioma, lung cancer, and others [76,77]. In addition, treatment with CAR T cells using the third-generation CARs that were designed based on the NZ-1 antibody also exhibited prolonged duration of survival using the xenograft models of glioblastomas [78]. This way, the efficacy demonstrated by treatments that target PDPN has been verified in both in vitro experiments and in xenograft models.
As stated above, the issue that requires most consideration for the clinical application of CAR T cell therapies is on-target off-tumor toxicity. In other words, for this treatment method, it is extremely important to select molecules specifically expressed in cancer cells. PDPN is highly expressed in various intractable tumors, such as lung cancer, malignant mesothelioma, malignant brain tumors, and esophageal cancer; however, it is also known to be expressed in normal cells, including lymphatic endothelial cells, alveolar epithelial cells, skin basal cells, and renal epithelial cells. Consequently, it is difficult to clinically apply CAR T cell therapies that are designed based on the anti-PDPN antibodies, such as NZ-1. However, in the study conducted by Kato and Kaneko, the authors succeeded in developing a cancer-specific antibody, which is known as the cancer-specific monoclonal antibody (CasMab), thereby adding a sense of feasibility to the actual use of CAR T cell therapies that can specifically target PDPN [79]. They focused on the PDPN molecule that is specifically expressed in the human glioblastoma cell line LN229, which had a different glycosylation pattern compared to the normal cells, and thereafter established CasMab by transplanting LN229 cells expressing PDPN (LN229/hPDPN) into mice. Among the established clones, a CasMab clone, LpMab-2, recognized human PDPN-expressing CHO cells (CHO/hPDPN), N-glycan deficient CHO/hPDPN cells, and LN229/hPDPN cells, whereas the clone did not recognize sialic acid-deficient CHO/hPDPN cells and O-glycan deficient CHO/hPDPN cells. These results indicate that the epitope recognized by LpMab-2 includes sialylated O-glycans. Furthermore, the authors determined the LpMab-2 target sequence by using deletion and point mutants of PDPN. As a result of these experiments, they concluded that the important epitope for LpMab-2 is the glycopeptide Thr55-Leu64. CasMab is an antibody that recognizes both the cancer-specific glycosylation and the PDPN-derived peptide sequence. Although it reacts with PDPN expressed by the cancer cells, it has been shown to be unreactive to PDPN expressed by the normal cells. Consequently, these CasMab and their scFv are promising and can be considered for application in PDPN-targeted therapy.
6. Conclusions and Perspectives
As discussed in this review article, if one can prepare CARs that maintain CasMab’s antigen specificity, it might be used to prevent the major issue of CAR T cell therapy, i.e., on-target off-tumor toxicity. In that case, this technology will change PDPN into an attractive target for CAR T cell therapy. There are, however, some concerns that should be considered when applying the technology to clinical practice. Although Kato and Kaneko have previously demonstrated that CasMab is a specific antibody directed against aberrantly glycosylated PDPN expressed in some human cancer cell lines and some patient-derived tumors, the number of patients’ samples is very limited. Therefore, more extensive studies are required to further confirm the antigen specificity of CasMab and the applicability of this concept to glioblastoma and other types of cancer. Another concern is the implications of tumor heterogeneity. Since CasMab recognizes both specific glycosylation and a PDPN-derived peptide as described above, it is assumed that the effectiveness of CasMab-based therapies is affected not only by the expression level of PDPN but also by the glycosylation pattern of PDPN. Therefore, a strategy targeting multiple antigens, including cancer-specific glycosylation patterns, may be useful to improve the therapeutic effect.
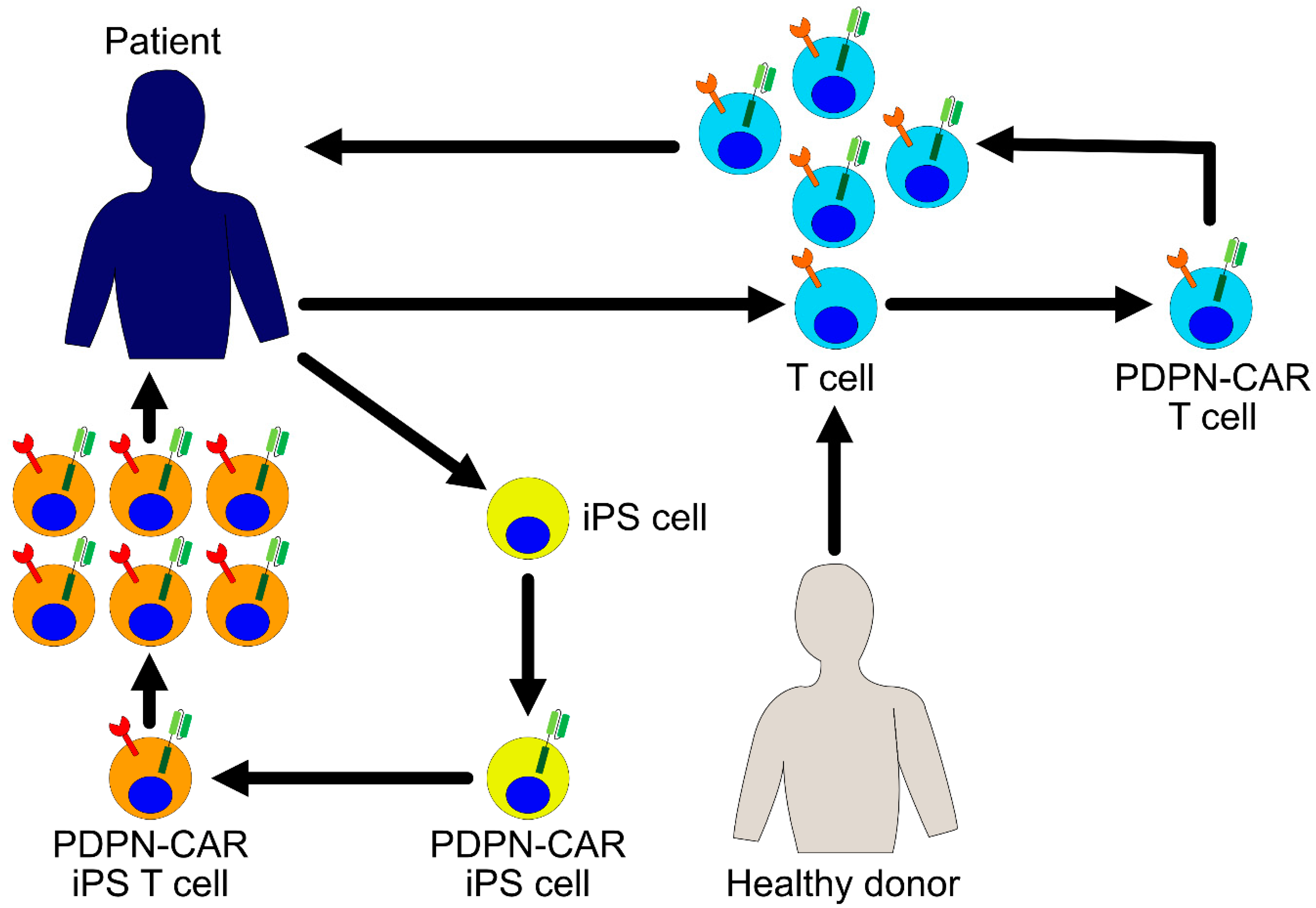
In the research studies that have been conducted so far, it has been found that long-term exposure to antigen stimulation and inflammatory signals in chronic diseases, such as cancer and viral infection, causes T cells to enter a state of exhaustion [80]. Exhausted T cells start expressing various inhibitory receptors and gradually lose their proliferation, cytokine production, and cytotoxic potential. Therefore, when the autologous T cells are used in the treatment, there is a concern that even while using the T cells harboring CARs, the proportion of exhausted T cells might affect the efficacy of the treatment. One approach for resolving this issue is developing immunity by renewing treatments that are focused on iPS cell techniques. Previously, we and other groups succeeded in inducing CTL differentiation using the iPS cells, and these regenerated CTLs have been found to exhibit considerable proliferation and cytokine production potential [81,82,83]. Moreover, iPS cells are efficiently transduced by recombinant lentiviral vectors and it is easy to control the quality of the gene-modified iPS cells. In the future, it is possible that the regenerated T cells derived from the iPS cells will become a platform for the expression of CARs and will become an important focus of research. The integration of this research is promising in asserting the development of PDPN-targeting CAR T cell therapies with considerable clinical efficacy for future studies (Figure 2).
Author Contributions
Writing and discussion, M.W. and S.K.; figure design, M.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Japan Society for the Promotion of Science (JSPS) KAKENHI, Grant Number 19K16862.
Acknowledgments
We would like to thank Yukinari Kato for providing anti-PDPN scFvs.
Conflicts of Interest
S.K. is a founder, shareholder, and chief scientific officer at Thyas Co., Ltd. and received research funding from Takeda Pharmaceutical Co., Ltd., Kirin Holdings Co., Ltd., Terumo Corp., Tosoh Corp., and Thyas Co., Ltd. M.W. received research funding from Takeda Pharmaceutical Co., Ltd.
References
- Peschon, J.J.; Morrissey, P.J.; Grabstein, K.H.; Ramsdell, F.J.; Maraskovsky, E.; Gliniak, B.C.; Park, L.S.; Ziegler, S.F.; Williams, D.E.; Ware, C.B.; et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J. Exp. Med. 1994, 180, 1955–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hozumi, K.; Mailhos, C.; Negishi, N.; Hirano, K.-I.; Yahata, T.; Ando, K.; Zuklys, S.; Holländer, G.A.; Shima, D.T.; Habu, S. Delta-like 4 is indispensable in thymic environment specific for T cell development. J. Exp. Med. 2008, 205, 2507–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, T.; Leisegang, M.; Park, J.-H.; Ren, L.; Kato, T.; Ikeda, Y.; Harada, M.; Kiyotani, K.; Lengyel, E.; Fleming, G.F.; et al. Induction of Neoantigen-Specific Cytotoxic T Cells and Construction of T-cell Receptor-Engineered T Cells for Ovarian Cancer. Clin. Cancer Res. 2018, 24, 5357–5367. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Brentjens, R.J.; Latouche, J.-B.; Santos, E.; Marti, F.; Gong, M.C.; Lyddane, C.; King, P.D.; Larson, S.; Weiss, M.; Rivière, I.; et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. 2003, 9, 279–286. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Finney, H.M.; Akbar, A.N.; Lawson, A.D.G. Activation of resting human primary T cells with chimeric receptors: Costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J. Immunol. 2004, 172, 104–113. [Google Scholar] [CrossRef]
- Kowolik, C.M.; Topp, M.S.; Gonzalez, S.; Pfeiffer, T.; Olivares, S.; Gonzalez, N.; Smith, D.D.; Forman, S.J.; Jensen, M.C.; Cooper, L.J.N. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006, 66, 10995–11004. [Google Scholar] [CrossRef] [Green Version]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Song, D.-G.; Ye, Q.; Poussin, M.; Harms, G.M.; Figini, M.; Powell, D.J., Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012, 119, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef] [Green Version]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef]
- Niedermeyer, J.; Scanlan, M.J.; Garin-Chesa, P.; Daiber, C.; Fiebig, H.H.; Old, L.J.; Rettig, W.J.; Schnapp, A. Mouse fibroblast activation protein: Molecular cloning, alternative splicing and expression in the reactive stroma of epithelial cancers. Int. J. Cancer 1997, 71, 383–389. [Google Scholar] [CrossRef]
- Rettig, W.J.; Garin-Chesa, P.; Beresford, H.R.; Oettgen, H.F.; Melamed, M.R.; Old, L.J. Cell-surface glycoproteins of human sarcomas: Differential expression in normal and malignant tissues and cultured cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3110–3114. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-C.S.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Zah, E.; Lin, M.-Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, H.; Ramakrishna, S.; Nguyen, S.; Fountaine, T.J.; Ponduri, A.; Stetler-Stevenson, M.; Yuan, C.M.; Haso, W.; Shern, J.F.; Shah, N.N.; et al. Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD22. Mol. Ther. Oncolytics 2018, 11, 127–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Zuo, S.; Deng, B.; Xu, X.; Li, C.; Zheng, Q.; Ling, Z.; Song, W.; Xu, J.; Duan, J.; et al. Sequential CD19-22 CAR T therapy induces sustained remission in children with r/r B-ALL. Blood 2020, 135, 387–391. [Google Scholar] [CrossRef]
- Tamada, K.; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin. Cancer Res. 2012, 18, 6436–6445. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A. Progress in human tumour immunology and immunotherapy. Nature 2001, 411, 380–384. [Google Scholar] [CrossRef]
- Refaeli, Y.; Van Parijs, L.; London, C.A.; Tschopp, J.; Abbas, A.K. Biochemical Mechanisms of IL-2–Regulated Fas-Mediated T Cell Apoptosis. Immunity 1998, 8, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Ku, C.C.; Murakami, M.; Sakamoto, A.; Kappler, J.; Marrack, P. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science 2000, 288, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA 1994, 271, 907–913. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Finkelstein, S.E.; Surman, D.R.; Lichtman, M.K.; Gattinoni, L.; Theoret, M.R.; Grewal, N.; Spiess, P.J.; Antony, P.A.; Palmer, D.C.; et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1969–1974. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.P.; Kovar, M.; Purton, J.F.; Cho, J.-H.; Boyman, O.; Surh, C.D.; Sprent, J. Converting IL-15 to a superagonist by binding to soluble IL-15R{alpha}. Proc. Natl. Acad. Sci. USA 2006, 103, 9166–9171. [Google Scholar] [CrossRef] [Green Version]
- Sato, N.; Patel, H.J.; Waldmann, T.A.; Tagaya, Y. The IL-15/IL-15Rα on cell surfaces enables sustained IL-15 activity and contributes to the long survival of CD8 memory T cells. Proc. Natl. Acad. Sci. USA 2007, 104, 588–593. [Google Scholar] [CrossRef] [Green Version]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.-F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.-A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef] [Green Version]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, K.A.; Hanafi, L.-A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef]
- Pober, J.S. Warner-Lambert/Parke-Davis award lecture. Cytokine-mediated activation of vascular endothelium. Physiology and pathology. Am. J. Pathol. 1988, 133, 426–433. [Google Scholar]
- Maruo, N.; Morita, I.; Shirao, M.; Murota, S. IL-6 increases endothelial permeability in vitro. Endocrinology 1992, 131, 710–714. [Google Scholar]
- Blecharz-Lang, K.G.; Wagner, J.; Fries, A.; Nieminen-Kelhä, M.; Rösner, J.; Schneider, U.C.; Vajkoczy, P. Interleukin 6-Mediated Endothelial Barrier Disturbances Can Be Attenuated by Blockade of the IL6 Receptor Expressed in Brain Microvascular Endothelial Cells. Transl. Stroke Res. 2018, 9, 631–642. [Google Scholar] [CrossRef]
- Torre, M.; Solomon, I.H.; Sutherland, C.L.; Nikiforow, S.; DeAngelo, D.J.; Stone, R.M.; Vaitkevicius, H.; Galinsky, I.A.; Padera, R.F.; Trede, N.; et al. Neuropathology of a Case With Fatal CAR T-Cell-Associated Cerebral Edema. J. Neuropathol. Exp. Neurol. 2018, 77, 877–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, K.A.; Turtle, C.J. Chimeric Antigen Receptor (CAR) T Cells: Lessons Learned from Targeting of CD19 in B-Cell Malignancies. Drugs 2017, 77, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Thistlethwaite, F.C.; Gilham, D.E.; Guest, R.D.; Rothwell, D.G.; Pillai, M.; Burt, D.J.; Byatte, A.J.; Kirillova, N.; Valle, J.W.; Sharma, S.K.; et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017, 66, 1425–1436. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef]
- Wang, X.; Chang, W.-C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef]
- Tasian, S.K.; Kenderian, S.S.; Shen, F.; Ruella, M.; Shestova, O.; Kozlowski, M.; Li, Y.; Schrank-Hacker, A.; Morrissette, J.J.D.; Carroll, M.; et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood 2017, 129, 2395–2407. [Google Scholar] [CrossRef] [Green Version]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [Green Version]
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [Green Version]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11, eaau5907. [Google Scholar] [CrossRef]
- Astarita, J.L.; Acton, S.E.; Turley, S.J. Podoplanin: Emerging functions in development, the immune system, and cancer. Front. Immunol. 2012, 3, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, H.; Retzbach, E.P.; Ramirez, M.I.; Liu, T.; Li, H.; Miller, W.T.; Goldberg, G.S. PKA and CDK5 can phosphorylate specific serines on the intracellular domain of podoplanin (PDPN) to inhibit cell motility. Exp. Cell Res. 2015, 335, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Martín-Villar, E.; Megías, D.; Castel, S.; Yurrita, M.M.; Vilaró, S.; Quintanilla, M. Podoplanin binds ERM proteins to activate RhoA and promote epithelial-mesenchymal transition. J. Cell Sci. 2006, 119, 4541–4553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicki, A.; Lehembre, F.; Wick, N.; Hantusch, B.; Kerjaschki, D.; Christofori, G. Tumor invasion in the absence of epithelial-mesenchymal transition: Podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell 2006, 9, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Fehon, R.G.; McClatchey, A.I.; Bretscher, A. Organizing the cell cortex: The role of ERM proteins. Nat. Rev. Mol. Cell Biol. 2010, 11, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Breiteneder-Geleff, S.; Matsui, K.; Soleiman, A.; Meraner, P.; Poczewski, H.; Kalt, R.; Schaffner, G.; Kerjaschki, D. Podoplanin, novel 43-kd membrane protein of glomerular epithelial cells, is down-regulated in puromycin nephrosis. Am. J. Pathol. 1997, 151, 1141–1152. [Google Scholar]
- Swartz, M.A.; Lund, A.W. Lymphatic and interstitial flow in the tumour microenvironment: Linking mechanobiology with immunity. Nat. Rev. Cancer 2012, 12, 210–219. [Google Scholar] [CrossRef]
- Kato, Y.; Kaneko, M.; Sata, M.; Fujita, N.; Tsuruo, T.; Osawa, M. Enhanced expression of Aggrus (T1alpha/podoplanin), a platelet-aggregation-inducing factor in lung squamous cell carcinoma. Tumour Biol. 2005, 26, 195–200. [Google Scholar] [CrossRef]
- Martín-Villar, E.; Scholl, F.G.; Gamallo, C.; Yurrita, M.M.; Muñoz-Guerra, M.; Cruces, J.; Quintanilla, M. Characterization of human PA2.26 antigen (T1alpha-2, podoplanin), a small membrane mucin induced in oral squamous cell carcinomas. Int. J. Cancer 2005, 113, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Kimura, N.; Kimura, I. Podoplanin as a marker for mesothelioma. Pathol. Int. 2005, 55, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Ordóñez, N.G. D2-40 and podoplanin are highly specific and sensitive immunohistochemical markers of epithelioid malignant mesothelioma. Hum. Pathol. 2005, 36, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Kato, Y.; Kaneko, M.K.; Nishikawa, R.; Hirose, T.; Matsutani, M. Increased expression of podoplanin in malignant astrocytic tumors as a novel molecular marker of malignant progression. Acta Neuropathol. 2006, 111, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Kunita, A.; Kashima, T.G.; Morishita, Y.; Fukayama, M.; Kato, Y.; Tsuruo, T.; Fujita, N. The platelet aggregation-inducing factor aggrus/podoplanin promotes pulmonary metastasis. Am. J. Pathol. 2007, 170, 1337–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemoto, A.; Miyata, K.; Fujita, N. Platelet-activating factor podoplanin: From discovery to drug development. Cancer Metastasis Rev. 2017, 36, 225–234. [Google Scholar] [CrossRef]
- Kato, Y.; Kaneko, M.K.; Kuno, A.; Uchiyama, N.; Amano, K.; Chiba, Y.; Hasegawa, Y.; Hirabayashi, J.; Narimatsu, H.; Mishima, K.; et al. Inhibition of tumor cell-induced platelet aggregation using a novel anti-podoplanin antibody reacting with its platelet-aggregation-stimulating domain. Biochem. Biophys. Res. Commun. 2006, 349, 1301–1307. [Google Scholar] [CrossRef]
- Abe, S.; Kaneko, M.K.; Tsuchihashi, Y.; Izumi, T.; Ogasawara, S.; Okada, N.; Sato, C.; Tobiume, M.; Otsuka, K.; Miyamoto, L.; et al. Antitumor effect of novel anti-podoplanin antibody NZ-12 against malignant pleural mesothelioma in an orthotopic xenograft model. Cancer Sci. 2016, 107, 1198–1205. [Google Scholar] [CrossRef]
- Shiina, S.; Ohno, M.; Ohka, F.; Kuramitsu, S.; Yamamichi, A.; Kato, A.; Motomura, K.; Tanahashi, K.; Yamamoto, T.; Watanabe, R.; et al. CAR T Cells Targeting Podoplanin Reduce Orthotopic Glioblastomas in Mouse Brains. Cancer Immunol Res. 2016, 4, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Kaneko, M.K. A cancer-specific monoclonal antibody recognizes the aberrantly glycosylated podoplanin. Sci. Rep. 2014, 4, 5924. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.-I.; Koseki, H.; Kawamoto, H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic structure of the three generations of CARs. CAR is a fusion protein composed of an extracellular scFv that is derived from VH and VL regions of an antibody, a transmembrane domain and intracellular signaling domains derived from T cell signaling proteins. First-generation CAR contains only CD3ζ chain for signal transduction. In addition to the CD3ζ chain, second-generation CAR possesses one of the costimulatory receptor-derived signal transduction domains, such as from CD28 and 4-1BB. Third-generation CAR consists of two costimulatory domains. CAR, chimeric antigen receptor; scFv, single chain variable fragment; VH, variable heavy chain; VL, variable light chain.
Figure 1.
Schematic structure of the three generations of CARs. CAR is a fusion protein composed of an extracellular scFv that is derived from VH and VL regions of an antibody, a transmembrane domain and intracellular signaling domains derived from T cell signaling proteins. First-generation CAR contains only CD3ζ chain for signal transduction. In addition to the CD3ζ chain, second-generation CAR possesses one of the costimulatory receptor-derived signal transduction domains, such as from CD28 and 4-1BB. Third-generation CAR consists of two costimulatory domains. CAR, chimeric antigen receptor; scFv, single chain variable fragment; VH, variable heavy chain; VL, variable light chain.

Figure 2.
PDPN-CAR expressing T cells for cancer immunotherapy. Patient derived- or healthy donor derived-T cells are harvested and transduced with a PDPN-CAR encoding gene ex vivo. The PDPN-CAR T cells are further expanded ex vivo and infused into the patient. Another possible approach is to use induced pluripotent stem (iPS) cells as a source of T cells. iPS cells are established from patient’s somatic cells and transduced with a PDPN-CAR ex vivo. PDPN-CAR expressing T cells are differentiated from these iPS cells, expanded, and infused into the patient.
Figure 2.
PDPN-CAR expressing T cells for cancer immunotherapy. Patient derived- or healthy donor derived-T cells are harvested and transduced with a PDPN-CAR encoding gene ex vivo. The PDPN-CAR T cells are further expanded ex vivo and infused into the patient. Another possible approach is to use induced pluripotent stem (iPS) cells as a source of T cells. iPS cells are established from patient’s somatic cells and transduced with a PDPN-CAR ex vivo. PDPN-CAR expressing T cells are differentiated from these iPS cells, expanded, and infused into the patient.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Waseda, M.; Kaneko, S. Podoplanin as an Attractive Target of CAR T Cell Therapy. Cells 2020, 9, 1971. https://doi.org/10.3390/cells9091971
AMA Style
Waseda M, Kaneko S. Podoplanin as an Attractive Target of CAR T Cell Therapy. Cells. 2020; 9(9):1971. https://doi.org/10.3390/cells9091971
Chicago/Turabian StyleWaseda, Masazumi, and Shin Kaneko. 2020. "Podoplanin as an Attractive Target of CAR T Cell Therapy" Cells 9, no. 9: 1971. https://doi.org/10.3390/cells9091971
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.