Mucopolysaccharidosis Type I: A Review of the Natural History and Molecular Pathology

 ,
,
Abstract
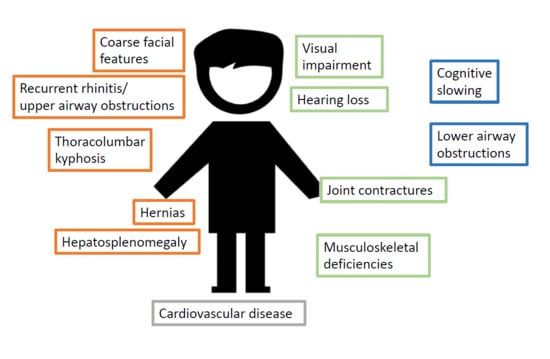
:
1. Introduction
2. Symptoms during the First 6 Months of Life
2.1. Facial Features
2.2. Abdominal Hernias
2.3. Hepatosplenomegaly
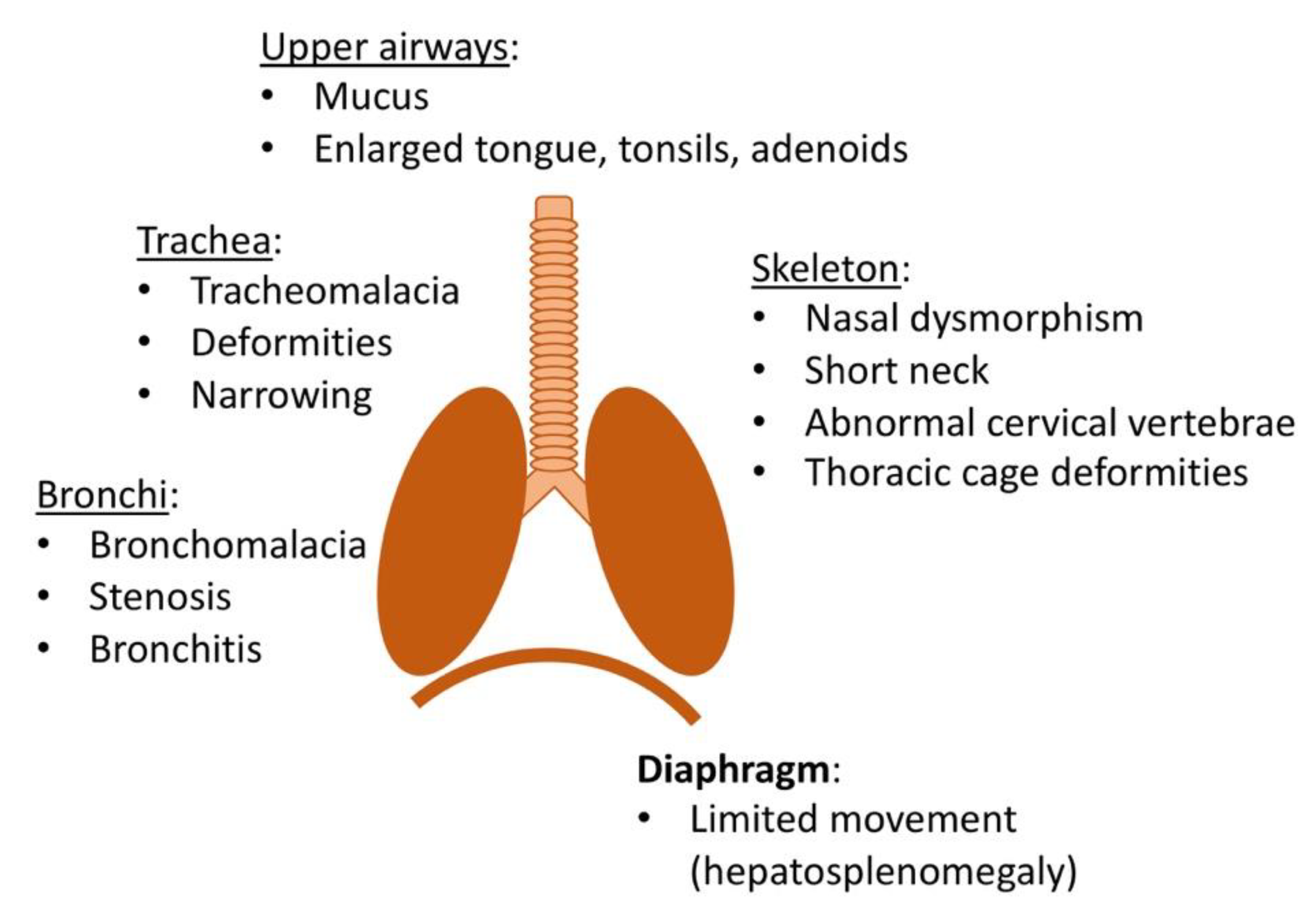
2.4. Respiratory and Pulmonary Manifestations
2.5. Diagnostic Tests of Respiratory Function
3. Symptoms Developing after 6 Months of Age
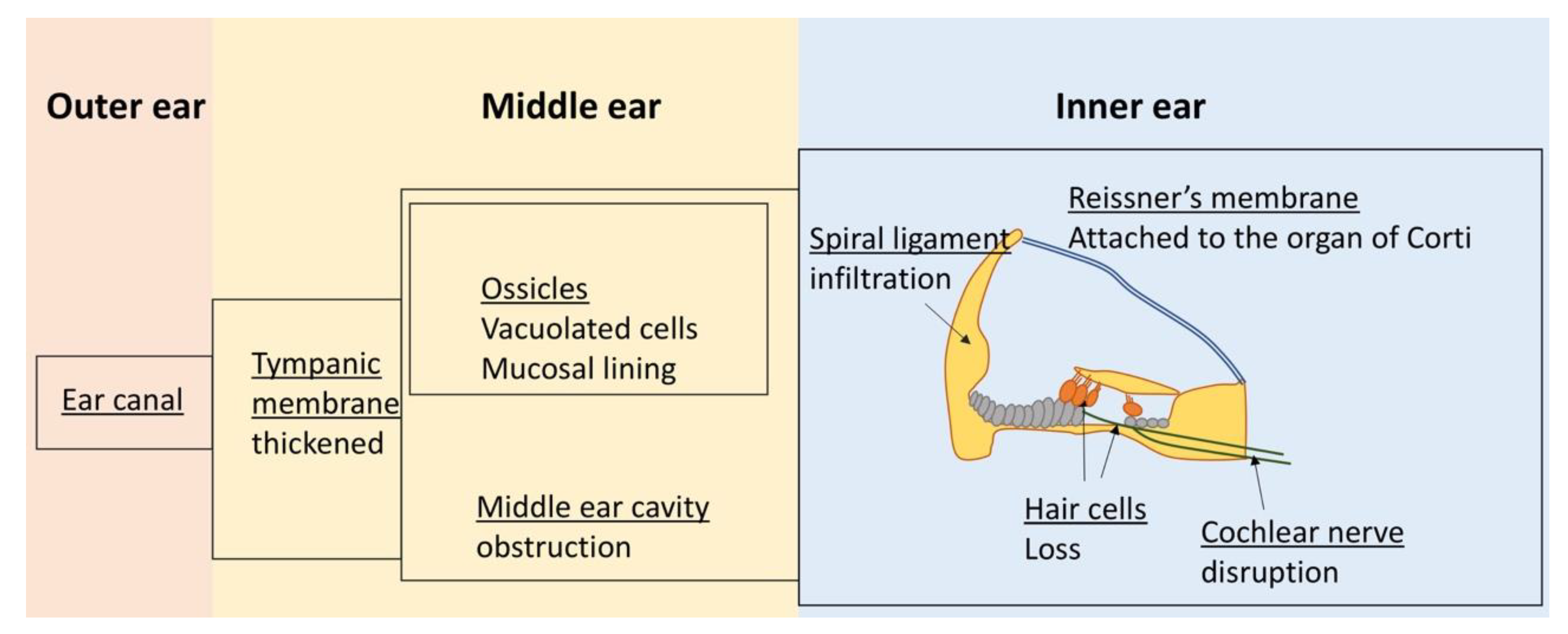
3.1. Hearing Loss
Diagnostics for Auditory Manifestations
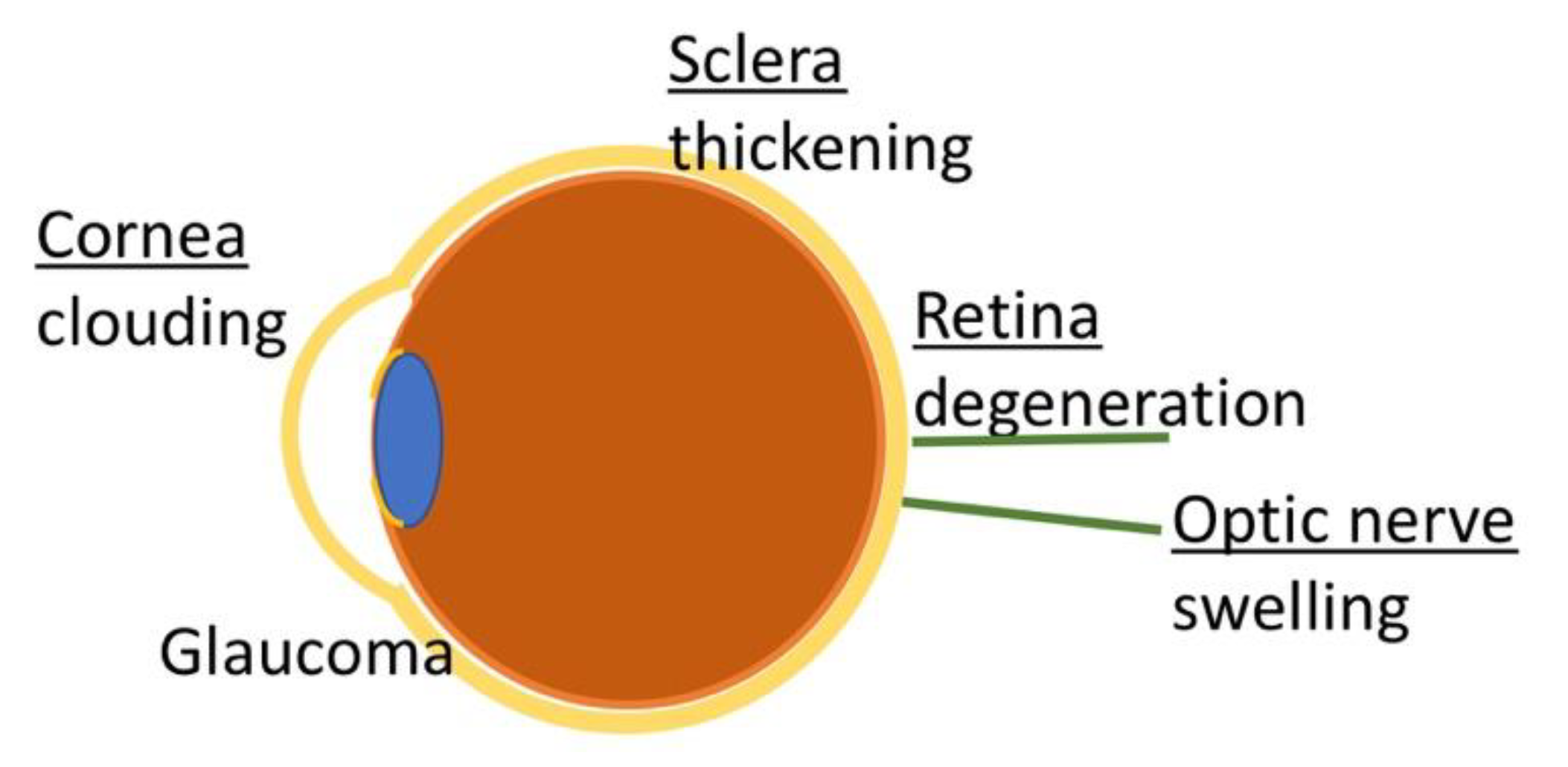
3.2. Ocular Manifestations
3.2.1. Corneal Clouding
3.2.2. Optic Nerve Swelling
3.2.3. Retinopathy
3.2.4. Glaucoma
3.2.5. Diagnostics for Ocular Manifestations
4. Skeletal Disease and Joint Symptoms in MPS I
Diagnostic Tests for Skeletal Deficiencies and Joint Symptoms
5. Symptoms Emerging after 1 Year of Life
5.1. Cognitive Impairment
5.2. Diagnostic Tests for Cognitive and Adaptive Skills
6. Cardiac Manifestations
6.1. Valve Abnormalities
6.2. Coronary Artery Disease
6.3. Other Vascular Changes
6.4. Diagnostics for Cardiac Manifestations
6.5. Cardiac Manifestations in Animal Models
7. Pathogenic Mechanisms
7.1. Cellular Pathology
7.2. Inflammatory Immune Responses
7.3. Biochemical Pathology
7.4. Biochemical Mechanism of Loss of Cardiac Elasticity
8. Conclusions
Funding
Conflicts of Interest
References
- Campos, D.; Monaga, M. Mucopolysaccharidosis type I: Current knowledge on its pathophysiological mechanisms. Metab. Brain Dis. 2012, 27, 121–129. [Google Scholar] [CrossRef]
- Beck, M.; Arn, P.; Giugliani, R.; Muenzer, J.; Okuyama, T.; Taylor, J.; Fallet, S. The natural history of MPS I: Global perspectives from the MPS I Registry. Genet. Med. 2014, 16, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Kiely, B.T.; Kohler, J.L.; Coletti, H.Y.; Poe, M.D.; Escolar, M.L. Early disease progression of Hurler syndrome. Orphanet J. Rare Dis. 2017, 12, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantopoulos, G.; Mccomb, R.D.; Dekaban, A.S. Neurochemistry of the Mucopolysaccharidoses: Brain Glycosaminoglycans in Normals and Four Types of Mucopolysaccharidoses. J. Neurochem. 1976, 26, 901–908. [Google Scholar] [CrossRef]
- Ikeno, T.; Minami, R.; Wagatsuma, K.; Fujibayashi, S.; Nakao, T.; Abo, K.; Tsugawa, S.; Taniguchi, S.; Takasago, Y. Prenatal diagnosis of Hurler’s syndrome-Biochemical studies on the affected fetus. Hum. Genet. 1981, 59, 353–359. [Google Scholar] [CrossRef]
- Crawfurd, M.A.; Dean, M.F.; Hunt, D.M.; Johnson, D.R.; MacDonald, R.R.; Muir, H.; Wright, E.A.; Wright, C.R. Early prenatal diagnosis of Hurler’s syndrome with termination of pregnancy and confirmatory findings on the fetus. J. Med Genet. 1973, 10, 144–153. [Google Scholar] [CrossRef]
- Bigger, B.W.; Begley, D.J.; Virgintino, D.; Pshezhetsky, A.V. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2018, 125, 322–331. [Google Scholar] [CrossRef]
- Jiang, Z.; Derrick-Roberts, A.L.K.; Jackson, M.R.; Rossouw, C.; Pyragius, C.E.; Xian, C.; Fletcher, J.; Byers, S. Delayed development of ossification centers in the tibia of prenatal and early postnatal MPS VII mice. Mol. Genet. Metab. 2018, 124, 135–142. [Google Scholar] [CrossRef]
- Cleary, M.; Wraith, J. The presenting features of mucopolysaccharidosis type IH (Hurler syndrome). Acta. Pædiatrica 1995, 84, 337–339. [Google Scholar] [CrossRef]
- Kuiper, G.A.; Meijer, O.L.M.; Langereis, E.J.; Wijburg, F.A. Failure to shorten the diagnostic delay in two ultra-orphan diseases (mucopolysaccharidosis types I and III): Potential causes and implications. Orphanet J. Rare Dis. 2018, 13, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Pastores, G.M.; Arn, P.; Beck, M.; Clarke, J.T.R.; Guffon, N.; Kaplan, P.; Muenzer, J.; Norato, D.Y.J.; Shapiro, E.; Thomas, J.; et al. The MPS I registry: Design, methodology, and early findings of a global disease registry for monitoring patients with Mucopolysaccharidosis Type I. Mol. Genet. Metab. 2007, 91, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Bruni, S.; Lavery, C.; Broomfield, A. The diagnostic journey of patients with mucopolysaccharidosis I: A real-world survey of patient and physician experiences. Mol. Genet. Metab. Rep. 2016, 8, 67–73. [Google Scholar] [CrossRef] [PubMed]
- De Ru, M.H.; Bouwman, M.G.; Wijburg, F.A.; van Zwieten, M.C.B. Experiences of parents and patients with the timing of Mucopolysaccharidosis type I (MPS I) diagnoses and its relevance to the ethical debate on newborn screening. Mol. Genet. Metab. 2012, 107, 501–507. [Google Scholar] [CrossRef]
- Colville, G.A.; Bax, M.A. Early presentation in the mucopolysaccharide disorders. Child Carehealth Dev. 1996, 22, 31–36. [Google Scholar] [CrossRef]
- Haskins, M.E.; Jezyk, P.F.; Desnick, R.J.; McDonough, S.K.; Patterson, D.F. Alpha-L-iduronidase deficiency in a cat: A model of mucopolysaccharidosis I. Pediatr. Res. 1979, 13, 1294–1297. [Google Scholar] [CrossRef] [Green Version]
- Traas, A.M.; Wang, P.; Ma, X.; Tittiger, M.; Schaller, L.; O’donnell, P.; Sleeper, M.M.; Vite, C.; Herati, R.; Aguirre, G.D.; et al. Correction of clinical manifestations of canine mucopolysaccharidosis I with neonatal retroviral vector gene therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 1423–1431. [Google Scholar] [CrossRef]
- Dierenfeld, A.D.; McEntee, M.F.; Vogler, C.A.; Vite, C.H.; Chen, A.H.; Passage, M.; Le, S.; Shah, S.; Jens, J.K.; Snella, E.M.; et al. Replacing the enzyme alpha-L-iduronidase at birth ameliorates symptoms in the brain and periphery of dogs with mucopolysaccharidosis type I. Sci. Transl. Med. 2010, 2, 60ra89. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.A.; Russell, C.S.; Pownall, S.; Warrington, C.L.; Borowski, A.; Dimmick, J.E.; Toone, J.; Jirik, F.R. Murine mucopolysaccharidosis type I: Targeted disruption of the murine alpha-L-iduronidase gene. Hum. Mol. Genet. 1997, 6, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Kuber, S. Hernia Surgery Simplified. In Hernia Surgery Simplified; Jaypee Brothers Medical Publishers Ltd.: New Dehli, India, 2013; p. 39. [Google Scholar]
- Kim, C.; Seo, J.; Chung, Y.; Ji, H.J.; Lee, J.; Sohn, J.; Lee, B.; Jo, E.C. Comparative study of idursulfase beta and idursulfase in vitro and in vivo. J. Hum. Genet. 2017, 62, 167–174. [Google Scholar] [CrossRef]
- Simmons, M.A.; Bruce, I.A.; Penney, S.; Wraith, E.; Rothera, M.P. Otorhinolaryngological manifestations of the mucopolysaccharidoses. Int. J. Pediatr. Otorhinolaryngol. 2005, 69, 589–595. [Google Scholar] [CrossRef]
- Muhlebach, M.S.; Wooten, W.; Muenzer, J. Respiratory Manifestations in Mucopolysaccharidoses. Paediatr. Respir. Rev. 2011, 12, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Arn, P.; Bruce, I.A.; Wraith, J.E.; Travers, H.; Fallet, S. Airway-related symptoms and surgeries in patients with mucopolysaccharidosis I. Ann. Otol. Rhinol. Laryngol. 2015, 124, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solaiman, S.S.; Rifkin, D.S.; Rao, H. Sleep apnea in hurler syndrome: Looking beyond the upper airway. J. Clin. Sleep Med. 2016, 12, 1423–1424. [Google Scholar] [CrossRef] [Green Version]
- Shih, S.L.; Lee, Y.J.; Lin, S.P.; Sheu, C.Y.; Blickman, J.G. Airway changes in children with mucopolysaccharidoses: CT evaluation. Acta Radiol. 2002, 43, 40–43. [Google Scholar] [CrossRef]
- Peters, M.E.; Arya, S.; Langer, L.O.; Gilbert, E.F.; Carlson, R.; Adkins, W. Narrow trachea in mucopolysaccharidoses. Pediatr. Radiol. 1985, 15, 225–228. [Google Scholar] [CrossRef]
- Kampmann, C.; Wiethoff, C.; Huth, R.; Staatz, G.; Mengel, E.; Beck, M.; Gehring, S.; Mewes, T.; Abu-Tair, T. Management of Life-Threatening Tracheal Stenosis and Tracheomalacia in Patients with Mucopolysaccharidoses. Jimd. Rep. 2017, 33, 33–39. [Google Scholar] [PubMed] [Green Version]
- Valayannopoulos, V.; De Blic, J.; Mahlaoui, N.; Stos, B.; Jaubert, F.; Bonnet, D.; Fischer, A.; De Lonlay, P. Laronidase for cardiopulmonary disease in Hurler syndrome 12 years after bone marrow transplantation. Pediatrics 2010, 126. [Google Scholar] [CrossRef]
- Berger, K.; Fagondes, S.; Giugliani, R.; Hardy, K.; Lee, K.; McArdle, C.; Scarpa, M.; Tobin, M.; Ward, S.; Rapoport, D. Respiratory and sleep disorders in mucop olysaccharidosis. J. Inherit. Metab. Dis. 2013, 36, 201–210. [Google Scholar] [CrossRef]
- Schuh, R.S.; Gonzalez, E.A.; Tavares, A.M.V.; Seolin, B.G.; de Elias, L.S.; Vera, L.N.P.; Kubaski, F.; Poletto, E.; Giugliani, R.; Teixeira, H.F.; et al. Neonatal nonviral gene editing with the CRISPR/Cas9 system improves some cardiovascular, respiratory, and bone disease features of the mucopolysaccharidosis I phenotype in mice. Gene Ther. 2019. [Google Scholar] [CrossRef]
- Gönüldaş, B.; Yilmaz, T.; Sivri, H.S.; Güçer, K.Ş.; Kilinç, K.; Genç, G.A.; Kiliç, M.; Coşkun, T. Mucopolysaccharidosis: Otolaryngologic findings, obstructive sleep apnea and accumulation of glucosaminoglycans in lymphatic tissue of the upper airway. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 944–949. [Google Scholar] [CrossRef]
- Burki, N. Spirometry; Other Pulmonary Function Tests. J Fam. Pract. 1981, 12, 119–124. [Google Scholar] [PubMed]
- Lin, S.P.; Shih, S.C.; Chuang, C.K.; Lee, K.S.; Chen, M.R.; Niu, D.M.; Chiu, P.C.; Lin, S.J.; Lin, H.Y. Characterization of pulmonary function impairments in patients with mucopolysaccharidoses-Changes with age and treatment. Pediatr. Pulmonol. 2014, 49, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Motamed, M.; Thorne, S.; Narula, A. Treatment of otitis media with effusion in children with mucopolysaccharidoses. Int. J. Pediatr. Otorhinolaryngol. 2000, 53, 121–124. [Google Scholar] [CrossRef]
- Kariya, S.; Schachern, P.A.; Nishizaki, K.; Paparella, M.M.; Cureoglu, S. Inner ear changes in mucopolysaccharidosis type I/Hurler syndrome. Otol. Neurotol. 2012, 33, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- Spellacy, E.; Watts, R.W.; Friedmann, I.; Crow, J. Histopathological studies of the temporal bones in hurler’s disease [mucopolysaccharidosis (mps) IH]. J. Laryngol. Otol. 1985, 99, 29–42. [Google Scholar] [CrossRef]
- Schachern, P.A.; Shea, D.A.; Paparella, M.M. Mucopolysaccharidosis I-H (Hurler’s syndrome) and human temporal bone histopathology. Ann. Otol. Rhinol. Laryngol. 1984, 93, 65–69. [Google Scholar] [CrossRef]
- Kelemen, G. Hurler ’s Syndrome; the hearing organ. J. Laryngol Otol. 1966, 80, 791–803. [Google Scholar] [CrossRef]
- Paparella, M.M.; Shea, D.; Meyerhoff, W.L.; Goycoolea, M.V. Silent otitis media. Laryngoscope 1980, 90, 1089–1098. [Google Scholar] [CrossRef]
- Woitge, H.W.; Friedmann, B.; Suttner, S.; Farahmand, I.; Müller, M.; Schmidt-Gayk, H.; Baertsch, P.; Ziegler, R.; Seibel, M.J. Changes in bone turnover induced by aerobic and anaerobic exercise in young males. J. Bone Miner. Res. 1998, 13, 1797–1804. [Google Scholar] [CrossRef]
- Schachern, P.A.; Cureoglu, S.; Tsuprun, V.; Paparella, M.M.; Whitley, C. Age-related functional and histopathological changes of the ear in the MPS I mouse. Int. J. Pediatr. Otorhinolaryngol. 2007, 71, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, L.; Hennig, A.K.; Kovacs, A.; Fu, A.; Chung, S.; Lee, D.; Wang, B.; Herati, R.S.; Mosinger Ogilvie, J.; et al. Liver-directed neonatal gene therapy prevents cardiac, bone, ear, and eye disease in mucopolysaccharidosis I mice. Mol. Ther. 2005, 11, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Hequembourg, S.; Liberman, M.C. Spiral ligament pathology: A major aspect of age-related cochlear degeneration in C57BL/6 mice. Jaro J. Assoc. Res. Otolaryngol. 2001, 2, 118–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hordeaux, J.; Deniaud, J.; Bemelmans, I.; Bertrand, L.; Moreau, S.; Amiaud, J.; Wyers, M.; Cherel, Y.; Colle, M.A. Histopathologic changes of the ear in canine models of mucopolysaccharidosis types I and VII. Vet. Pathol. 2011, 48, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Shih, S.C.; Chuang, C.K.; Lee, K.S.; Chen, M.R.; Lin, H.C.; Chiu, P.C.; Niu, D.M.; Lin, S.P. Assessment of hearing loss by pure-tone audiometry in patients with mucopolysaccharidoses. Mol. Genet. Metab. 2014, 111, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Chimelo, F.T.; Silva, L.A.F.; Kim, C.A.; Matas, C.G. Audiological characteristics in mucopolysaccharidosis: A systematic literature review. Rev. Cefac. 2019, 21, 1–8. [Google Scholar] [CrossRef]
- Zanetti, D.; Vezzani, M.; Di Berardino, F.; Gasperini, S.; Parini, R. Characterization of Hearing Loss in Children with Mucopolysaccharidosis, An Excursus into Hearing Loss. InTechOpen 2018. [Google Scholar] [CrossRef] [Green Version]
- da Silveira, M.R.M.; Buriti, A.K.L.; Martins, A.M.; Gil, D.; de Azevedo, M.F. Audiometric evaluation in individuals with mucopolysaccharidosis. Clinics (Sao Paulobrazil) 2018, 73, e523. [Google Scholar] [CrossRef]
- Ashworth, J.L.; Biswas, S.; Wraith, E.; Lloyd, I.C. Mucopolysaccharidoses and the eye. Surv. Ophthalmol. 2006, 51, 1–17. [Google Scholar] [CrossRef]
- Tomatsu, S.; Pitz, S.; Hampel, U. Ophthalmological Findings in Mucopolysaccharidoses. J. Clin. Med. 2019, 8, 1467. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Pinsky, P.M. Mechanisms of self-organization for the collagen fibril lattice in the human cornea. J. R. Soc. Interface 2013, 10. [Google Scholar] [CrossRef]
- Alroy, J.; Haskins, M.; Birk, D.E. Altered corneal stromal matrix organization is associated with mucopolysaccharidosis I, III and VI. Exp. Eye Res. 1999, 68, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Bothun, E.; Hardten, D.; Tolar, J.; McLoon, L. A novel explanation of corneal clouding in a bone marrow transplant-treated patient with Hurler syndrome. Exp. Eye Res. 2016, 148, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashworth, J.L.; Biswas, S.; Wraith, E.; Lloyd, I.C. The ocular features of the mucopolysaccharidoses. Eye 2006, 20, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Sahenk, Z.; Al-Zaidy, S.; Rodino-Klapac, L.R.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Miller, N.; Yalvac, M.; Dvorchik, I.; et al. Follistatin Gene Therapy for Sporadic Inclusion Body Myositis Improves Functional Outcomes. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Haskins, M.E.; Aguirre, G.D.; Jezyk, P.F.; Desnick, R.J.; Patterson, D.F. The Pathology of the Feline Model of Mucopolysaccharidosis I. Am. J. Pathol. 1983, 112, 27–36. [Google Scholar] [CrossRef]
- Constantopoulos, G.; Scott, J.A.; Shull, R.M. Corneal opacity in canine MPS I. Changes after bone marrow transplantation. Investig. Ophthalmol. Vis. Sci. 1989, 30, 1802–1807. [Google Scholar]
- Gonzalez, E.A.; Visioli, F.; Pasqualim, G.; de Souza, C.F.M.; Marinho, D.R.; Giugliani, R.; Matte, U.; Baldo, G. Progressive eye pathology in mucopolysaccharidosis type I mice and effects of enzyme replacement therapy. Clin. Exp. Ophthalmol. 2020, 48, 334–342. [Google Scholar] [CrossRef]
- Collins, M.L.Z.; Traboulsi, E.I.; Maumenee, I.H. Optic Nerve Head Swelling and Optic Atrophy in the Systemic Mucopolysaccharides. Ophthalmology 1990, 97, 1445–1449. [Google Scholar] [CrossRef]
- Lin, H.Y.; Chan, W.C.; Chen, L.J.; Lee, Y.C.; Yeh, S.I.; Niu, D.M.; Chiu, P.C.; Tsai, W.H.; Hwu, W.L.; Chuang, C.K.; et al. Ophthalmologic manifestations in Taiwanese patients with mucopolysaccharidoses. Mol. Genet. Genom. Med. 2019, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gratton, S.M.; Neerukonda, T. Mucopolysaccharidosis Type I and Bilateral Optic Disc Edema. Neuro-Ophthalmol. 2019, 43, 394–396. [Google Scholar] [CrossRef]
- Schumacher, R.G.; Brzezinska, R.; Schulze-Frenking, G.; Pitz, S. Sonographic ocular findings in patients with mucopolysaccharidoses I, II and VI. Pediatr. Radiol. 2008, 38, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, A.; Bruwer, Z.; Al-Thihli, K. An update on ocular involvement in mucopolysaccharidoses. Curr. Opin. Ophthalmol. 2013, 24, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Gills, J.P.; Hobson, R.; Hanley, W.B.; McKuskick, V. Electroretinography and Fundus Oculi Findings in Hurler’s Disease and Allied Mucopolysaccharidoses. Arch Ophthalmol. 1965, 74, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Jensen, O.A.; Pedersen, C.; Schwartz, M.; Vestermark, S.; Warburg, M. Hurler/Scheie phenotype. Report of an inbred sibship with tapeto-retinal degeneration and electron-microscopie examination of the conjuctiva. Ophthalmologica 1978, 176, 194–204. [Google Scholar] [CrossRef]
- Sornalingam, K.; Javed, A.; Aslam, T.; Sergouniotis, P.; Jones, S.; Ghosh, A.; Ashworth, J. Variability in the ocular phenotype in mucopolysaccharidosis. Br. J. Ophthalmol. 2019, 103, 504–510. [Google Scholar] [CrossRef]
- Seok, S.; Lyu, I.J.; Park, K.A.; Oh, S.Y. Spectral domain optical coherence tomography imaging of mucopolysaccharidoses I, II, and VI A. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 2111–2119. [Google Scholar] [CrossRef]
- Newkirk, K.M.; Atkins, R.M.; Dickson, P.I.; Rohrbach, B.W.; McEntee, M.F. Ocular lesions in canine mucopolysaccharidosis I and response to enzyme replacement therapy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5130–5135. [Google Scholar] [CrossRef]
- Summers, C.G.; Ashworth, J.L. Ocular manifestations as key features for diagnosing mucopolysaccharidoses. Rheumatology 2011, 50, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Nowaczyk, M.J.; Clarke, J.T.R.; Morin, J.D. Glaucoma as an early complication of Hurler’s disease. Arch. Dis. Child. 1988, 63, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Spellacy, E.; Bankes, J.L.K.; Crow, J.; Dourmashkin, R.; Shah, D.; Watts, R.W. Glaucoma in a case of Hurler disease. Br. J. Ophthalmol. 1980, 64, 773–778. [Google Scholar] [CrossRef]
- Del Longo, A.; Piozzi, E.; Schweizer, F. Ocular features in mucopolysaccharidosis: Diagnosis and treatment. Ital. J. Pediatr. 2018, 44. [Google Scholar] [CrossRef] [PubMed]
- Fahnehjelm, K.T.; Ashworth, J.L.; Pitz, S.; Olsson, M.; Törnquist, A.L.; Lindahl, P.; Summers, C.G. Clinical guidelines for diagnosing and managing ocular manifestations in children with mucopolysaccharidosis. Acta Ophthalmol. 2012, 90, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Wasielica-Poslednik, J.; Butsch, C.; Lampe, C.; Elflein, H.; Lamparter, J.; Weyer, V.; Pitz, S. Comparison of rebound tonometry, perkins applanation tonometry and ocular response analyser in mucopolysaccharidosis patients. PLoS ONE 2015, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Javed, A.; Aslam, T.; Ashworth, J. Use of new imaging in detecting and monitoring ocular manifestations of the mucopolysaccharidoses. Acta Ophthalmol. 2016, 94, e676–e682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.K. Orthopaedic aspects of mucopolysaccharidoses. Rheumatology 2011, 50, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Polgreen, L.E.; Tolar, J.; Plog, M.; Himes, J.H.; Orchard, P.J.; Whitley, C.B.; Miller, B.S.; Petryk, A. Growth and endocrine function in patients with Hurler syndrome after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2008, 41, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Polgreen, L.E.; Lund, T.C.; Braunlin, E.; Tolar, J.; Miller, B.S.; Fung, E.; Whitley, C.B.; Eisengart, J.B.; Northrop, E.; Rudser, K.; et al. Clinical trial of laronidase in Hurler syndrome after hematopoietic cell transplantation. Pediatr. Res. 2019. [Google Scholar] [CrossRef]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O’Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: An international multicenter study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef] [Green Version]
- Weisstein, J.S.; Delgado, E.; Steinbach, L.S.; Hart, K.; Packman, S. Musculoskeletal manifestations of Hurler syndrome: Long-term follow-up after bone marrow transplantation. J. Pediatr. Orthop. 2004, 24, 97–101. [Google Scholar] [CrossRef]
- Viskochil, D.; Clarke, L.A.; Bay, L.; Keenan, H.; Muenzer, J.; Guffon, N. Growth patterns for untreated individuals with MPS I: Report from the international MPS I registry. Am. J. Med. Genet. Part A 2019, 179, 2425–2432. [Google Scholar] [CrossRef] [Green Version]
- Lachman, R.; Martin, K.W.; Castro, S.; Basto, M.A.; Adams, A.; Teles, E.L. Radiologic and neuroradiologic findings in the mucopolysaccharidoses. J. Pediatr. Rehabil. Med. 2010, 3, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandon, V.; Williamson, J.B.; Cowie, R.A.; Wraith, J.E. Spinal problems in mucopolysaccharidosis I (Hurler syndrome). J. Bone Jt. Surg. Br. 1996, 78, 938–944. [Google Scholar] [CrossRef]
- Belani, K.G.; Krivit, W.; Carpenter, B.L.; Braunlin, E.; Buckley, J.J.; Liao, J.C.; Floyd, T.; Leonard, A.S.; Summers, C.G.; Levine, S.; et al. Children with mucopolysaccharidosis: Perioperative care, morbidity, mortality, and new findings. J. Pediatr. Surg. 1993, 28, 403–410. [Google Scholar] [CrossRef]
- Hite, S.H.; Peters, C.; Krivit, W. Correction of odontoid dysplasia following bone-marrow transplantation and engraftment (in Hurler syndrome MPS 1H). Pediatr. Radiol. 2000, 30, 464–470. [Google Scholar] [CrossRef]
- Field, R.E.; Buchanan, J.A.; Copplemans, M.G.; Aichroth, P.M. Bone-marrow transplantation in Hurler’s syndrome. Effect on skeletal development. J. Bone Jt. Surg. Br. 1994, 76, 975–981. [Google Scholar] [CrossRef]
- Taylor, C.; Brady, P.; O’Meara, A.; Moore, D.; Dowling, F.; Fogarty, E. Mobility in Hurler syndrome. J. Pediatr. Orthop. 2008, 28, 163–168. [Google Scholar] [CrossRef]
- Clarke, L.A. Pathogenesis of skeletal; connective tissue involvement in the mucopolysaccharidoses: Glycosaminoglycan storage is merely the instigator. Rheumatology 2011, 50, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Guffon, N.; Journeau, P.; Brassier, A.; Leger, J.; Chevallier, B. Growth impairment and limited range of joint motion in children should raise suspicion of an attenuated form of mucopolysaccharidosis: expert opinion. Eur. J. Pediatr. 2019, 178, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Polgreen, L.E.; Thomas, W.; Fung, E.; Viskochil, D.; Stevenson, D.A.; Steinberger, J.; Orchard, P.; Whitley, C.B.; Ensrud, K.E. Low bone mineral content and challenges in interpretation of dual-energy X-ray absorptiometry in children with mucopolysaccharidosis types I, II, and VI. J. Clin. Densitom. 2014, 17, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Shih, S.C.; Chuang, C.K.; Chen, M.R.; Niu, D.M.; Lin, S.P. Assessment of bone mineral density by dual energy X-ray absorptiometry in patients with mucopolysaccharidoses. Orphanet J. Rare Dis. 2013, 8, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Breyer, S.; Löbel, U.; Yarar, S.; Stücker, R.; Ullrich, K.; Müller, I.; Muschol, N. Musculoskeletal manifestations in mucopolysaccharidosis type I (Hurler syndrome) following hematopoietic stem cell transplantation. Orphanet J. Rare Dis. 2016, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hufnagel, L.; Kreuger, J.; Cohen, S.M.; Shraiman, B.I. On the role of glypicans in the process of morphogen gradient formation. Dev. Biol. 2006, 300, 512–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billings, P.C.; Pacifici, M. Interactions of signaling proteins, growth factors and other proteins with heparan sulfate: Mechanisms and mysteries. Connect. Tissue Res. 2015, 56, 272–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silveri, C.P.; Kaplan, F.S.; Fallon, M.D.; Bayever, E.; August, C.S. Hurler syndrome with special reference to histologic abnormalities of the growth plate. Clin. Orthop. Relat. Res. 1991, 269, 305–311. [Google Scholar] [CrossRef]
- Silberberg, R.; Rimoin, D.; Rosenthal, R.; Hasler, M. Ultrastructure of cartilage in the Hurler and Sanfilippo syndromes. Arch. Pathol. 1972, 94, 500–510. [Google Scholar] [PubMed]
- Nogami, H.; Oohira, A.; Ozeki, K.; Oki, T.; Ogino, T.; Murachi, S. Ultrastructure of cartilage in heritable disorders of connective tissue. Clin. Orthop. Relat. Res. 1979, 251–259. [Google Scholar] [CrossRef]
- Pievani, A.; Azario, I.; Antolini, L.; Shimada, T.; Patel, P.; Remoli, C.; Rambaldi, B.; Valsecchi, M.; Riminucci, M.; Biondi, A.; et al. Neonatal bone marrow transplantation prevents bone pathology in a mouse model of mucopolysaccharidosis type I. Blood 2014, 124. [Google Scholar] [CrossRef]
- Oussoren, E.; Brands, M.M.M.G.; Ruijter, G.J.G.; van der Ploeg, A.T.; Reuser, A.J.J. Bone, joint and tooth development in mucopolysaccharidoses: Relevance to therapeutic options. Biochim. Et Biophys. Acta Mol. Basis Dis. 2011, 1812, 1542–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, C.; Hendson, G.; Jevon, G.; Matlock, T.; Yu, J.; Aklujkar, M.; Ng, K.-Y.; Clarke, L.A. Murine MPS I: Insights into the pathogenesis of Hurler syndrome. Clin. Genet. 2008, 53, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Heppner, J.M.; Zaucke, F.; Clarke, L.A. Extracellular matrix disruption is an early event in the pathogenesis of skeletal disease in mucopolysaccharidosis I. Mol. Genet. Metab. 2015, 114, 146–155. [Google Scholar] [CrossRef]
- Wilson, S.; Hashamiyan, S.; Clarke, L.; Saftig, P.; Mort, J.; Dejica, V.M.; Brömme, D. Glycosaminoglycan-mediated loss of cathepsin K collagenolytic activity in MPS I contributes to osteoclast and growth plate abnormalities. Am. J. Pathol. 2009, 175, 2053–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaro, J.A.; Baron, M.D.; del Alcazar, C.M.; O’Donnell, P.; Shore, E.M.; Elliott, D.M.; Ponder, K.P.; Haskins, M.E.; Smith, L.J. Postnatal progression of bone disease in the cervical spines of mucopolysaccharidosis I dogs. Bone 2013, 55, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonaro, C.M.; D’Angelo, M.; He, X.; Eliyahu, E.; Shtraizent, N.; Haskins, M.E.; Schuchman, E.H. Mechanism of glycosaminoglycan-mediated bone and joint disease: Implications for the mucopolysaccharidoses and other connective tissue diseases. Am. J. Pathol. 2008, 172, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonaro, C.M.; D’Angelo, M.; Haskins, M.E.; Schuchman, E.H. Joint and bone disease in mucopolysaccharidoses VI and VII: Identification of new therapeutic targets and biomarkers using animal models. Pediatr. Res. 2005, 57, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.A. Laronidase for the treatment of mucopolysaccharidosis type I. Expert Rev. Endocrinol. Metab. 2011, 6, 755–768. [Google Scholar] [CrossRef]
- Kirkpatrick, K.; Ellwood, J.; Walker, R.W. Mucopolysaccharidosis type I (Hurler syndrome) and anesthesia: The impact of bone marrow transplantation, enzyme replacement therapy, and fiberoptic intubation on airway management. Paediatric Anaesth 2012, 22, 745–751. [Google Scholar] [CrossRef]
- Shapiro, E.G.; Whitley, C.B.; Eisengart, J.B. Beneath the floor: Re-analysis of neurodevelopmental outcomes in untreated Hurler syndrome. Orphanet J. Rare Dis. 2018, 13, 76. [Google Scholar] [CrossRef]
- Aldenhoven, M.; Boelens, J.; de Koning, T.J. The Clinical Outcome of Hurler Syndrome after Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2008, 14, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Kunin-Batson, A.S.; Shapiro, E.G.; Rudser, K.D.; Lavery, C.A.; Bjoraker, K.J.; Jones, S.A.; Wynn, R.F.; Vellodi, A.; Tolar, J.; Orchard, P.J. Long-Term Cognitive and Functional Outcomes in Children with Mucopolysaccharidosis (MPS)-IH (Hurler Syndrome) Treated with Hematopoietic Cell Transplantation. Jimd. Rep. 2016, 29, 95–102. [Google Scholar]
- Shapiro, E.G.; Nestrasil, I.; Rudser, K.; Delaney, K.; Kovac, V.; Ahmed, A.; Yund, B.; Orchard, P.J.; Eisengart, J.; Niklason, G.R.; et al. Neurocognition across the spectrum of mucopolysaccharidosis type I: Age, severity, and treatment. Mol. Genet. Metab. 2015, 116, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Bjoraker, K.J.; Delaney, K.; Peters, C.; Krivit, W.; Shapiro, E.G. Long-term outcomes of adaptive functions for children with mucopolysaccharidosis I (Hurler syndrome) treated with hematopoietic stem cell transplantation. J. Dev. Behav. Pediatr. 2006, 27, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Pitt, C.; Lavery, C.; Wager, N. Psychosocial outcomes of bone marrow transplant for individuals affected by Mucopolysaccharidosis I Hurler Disease: Patient social competency. Child. Carehealth Dev. 2009, 35, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Zafeiriou, D.I.; Batzios, S.P. Brain and spinal MR imaging findings in mucopolysaccharidoses: A review. Am. J. Neuroradiol. 2013, 34, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufeld, E.F.; Muenzer, I. The Metabolic & Molecular Basis of Inherited Disease, 8th ed.; Scriver, C.R., Ed.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- De Pasquale, V.; Pavone, L.M. Heparan sulfate proteoglycans: The sweet side of development turns sour in mucopolysaccharidoses. Biochim. Et Biophys. Acta Mol. Basis Dis. 2019, 1865, 165539. [Google Scholar] [CrossRef]
- Fecarotta, S.; Tarallo, A.; Damiano, C.; Minopoli, N.; Parenti, G. Pathogenesis of mucopolysaccharidoses, an update. Int. J. Mol. Sci. 2020, 21, 2515. [Google Scholar] [CrossRef] [Green Version]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef]
- Gallagher, J.T. Multiprotein signalling complexes: Regional assembly on heparan sulphate. Biochem. Soc. Trans. 2006, 34, 438–441. [Google Scholar] [CrossRef]
- Holley, R.J.; Deligny, A.; Wei, W.; Watson, H.A.; Niñonuevo, M.R.; Dagälv, A.; Leary, J.A.; Bigger, B.W.; Kjellén, L.; Merry, C.L.R. Mucopolysaccharidosis type I, unique structure of accumulated heparan sulfate and increased N-sulfotransferase activity in mice lacking α-L-iduronidase. J. Biol. Chem. 2011, 286, 37515–37524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumkotter, J.; Cantz, M. Decreased ganglioside neuraminidase activity in fibroblasts from mucopolysaccharidosis patients. Inhibition of the activity in vitro by sulfated glycosaminoglycans and other compounds. Biochim. Biophys. Acta. 1983, 761, 163–170. [Google Scholar] [CrossRef]
- Kint, J.; Dacremont, G.; Carton, D.; Orye, E.; Hooft, C. Mucopolysaccharidosis: Secondarily Induced Abnormal. Science 1973, 181, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Constantopoulos, G.; Iqbal, K.; Dekaban, A.S. Mucopolysaccharidosis Types IH, IS, II and IIIA: Glycosaminoglycans and Lipids of Isolated Brain Cells and Other Fractions from Autopsied Tissues. J. Neurochem. 1980, 34, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, R.; Dobrenis, K.; Walkley, S.U. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J. Comp. Neurol. 2004, 480, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Nestrasil, I.; Shapiro, E.; Svatkova, A.; Dickson, P.; Chen, A.; Wakumoto, A.; Ahmed, A.; Stehel, E.; McNeil, S.; Gravance, C.; et al. Intrathecal enzyme replacement therapy reverses cognitive decline in mucopolysaccharidosis type I. Am. J. Med. Genet. A 2017, 173, 780–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, E.; Guler, O.E.; Rudser, K.; Delaney, K.; Bjoraker, K.; Whitley, C.; Tolar, J.; Orchard, P.; Provenzale, J.; Thomas, K.M. An exploratory study of brain function and structure in mucopolysaccharidosis type I: Long term observations following hematopoietic cell transplantation (HCT). Mol. Genet. Metab. 2012, 107, 116–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, K.E.; Rudser, K.D.; Nestrasil, I.; Kovac, V.; Delaney, K.A.; Wozniak, J.R.; Mueller, B.A.; Lim, K.O.; Eisengart, J.B.; Mamak, E.G.; et al. Attention and corpus callosum volumes in individuals with mucopolysaccharidosis type i. Neurology 2019, 92, E2321–E2328. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, E.; Huisman, T.A.G.M.; Boltshauser, E.; Scheer, I.; Güngör, T.; Tekes, A.; Maegawa, G.H.; Poretti, A. Mucopolysaccharidoses type I and II: New neuroimaging findings in the cerebellum. Eur. J. Paediatr. Neurol. 2014, 18, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Eisengart, J.B.; Rudser, K.D.; Xue, Y.; Orchard, P.; Miller, W.; Lund, T.; Van der Ploeg, A.; Mercer, J.; Jones, S.; Mengel, K.E.; et al. Long-term outcomes of systemic therapies for Hurler syndrome: An international multicenter comparison. Genet. Med. 2018, 20, 1423–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisengart, J.B.; Jarnes, J.; Ahmed, A.; Nestrasil, I.; Ziegler, R.; Delaney, K.; Shapiro, E.; Whitley, C. Long-term cognitive and somatic outcomes of enzyme replacement therapy in untransplanted Hurler syndrome. Mol. Genet. Metab. Rep. 2017, 13, 64–68. [Google Scholar] [CrossRef]
- Clarke, L.A.; Atherton, A.M.; Burton, B.K.; Day-Salvatore, D.L.; Kaplan, P.; Leslie, N.D.; Scott, C.R.; Stockton, D.W.; Thomas, J.A.; Muenzer, J. Mucopolysaccharidosis Type I Newborn Screening: Best Practices for Diagnosis and Management. J. Pediatr. 2017, 182, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef]
- Ohmi, K.; Greenberg, D.S.; Rajavel, K.S.; Ryazantsev, S.; Li, H.H.; Neufeld, E.F. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. USA 2003, 100, 1902–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkley, S.U.; Vanier, M. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta 2009, 1793, 726–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vite, C.H.; Nestrasil, I.; Mlikotic, A.; Jens, J.K.; Snella, E.; Gross, W.; Shapiro, E.G.; Kovac, V.; Provenzale, J.M.; Chen, S.; et al. Features of brain MRI in dogs with treated and untreated mucopolysaccharidosis type i. Comp. Med. 2013, 63, 163–173. [Google Scholar] [PubMed]
- Vite, C.H.; Wang, P.; Patel, R.T.; Walton, R.M.; Walkley, S.U.; Sellers, R.S.; Ellinwood, N.M.; Cheng, A.S.; White, J.T.; O’Neill, C.A.; et al. Biodistribution and pharmacodynamics of recombinant human alpha-L-iduronidase (rhIDU) in mucopolysaccharidosis type I-affected cats following multiple intrathecal administrations. Mol. Genet. Metab. 2011, 103, 268–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belur, L.R.; Temme, A.; Podetz-Pedersen, K.M.; Riedl, M.; Vulchanova, L.; Robinson, N.; Hanson, L.R.; Kozarsky, K.F.; Orchard, P.J.; Frey, W.H., 2nd; et al. Intranasal Adeno-Associated Virus Mediated Gene Delivery and Expression of Human Iduronidase in the Central Nervous System: A Noninvasive and Effective Approach for Prevention of Neurologic Disease in Mucopolysaccharidosis Type I. Hum. Gene Ther. 2017, 28, 576–587. [Google Scholar] [CrossRef]
- Pan, D.; Sciascia, A.; Vorhees, C.V.; Williams, M.T. Progression of multiple behavioral deficits with various ages of onset in a murine model of Hurler syndrome. Brain Res. 2008, 1188, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Hartung, S.D.; Frandsen, J.L.; Pan, D.; Koniar, B.L.; Graupman, P.; Gunther, R.; Low, W.C.; Whitley, C.B.; McIvor, R.S. Correction of metabolic, craniofacial, and neurologic abnormalities in MPS I mice treated at birth with adeno-associated virus vector transducing the human alpha-L-iduronidase gene. Mol. Ther. J. Am. Soc. Gene Ther. 2004, 9, 866–875. [Google Scholar] [CrossRef]
- Reolon, G.K.; Braga, L.M.E.; Camassola, M.; Luft, T.; Henriques, J.A.P.; Nardi, N.B.; Roesler, R. Long-term memory for aversive training is impaired in Idua-/- mice, a genetic model of mucopolysaccharidosis type I. Brain Res. 2006, 1076, 225–230. [Google Scholar] [CrossRef]
- Garcia-Rivera, M.F.; Colvin-Wanshura, L.E.; Nelson, M.S.; Nan, Z.; Khan, S.A.; Rogers, T.B.; Maitra, I.; Low, W.C.; Gupta, P. Characterization of an immunodeficient mouse model of mucopolysaccharidosis type I suitable for preclinical testing of human stem cell and gene therapy. Brain Res. Bull. 2007, 74, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Provoost, L.; Siracusa, C.; Stefanovski, D.; Che, Y.; Li, M.; Casal, M. Cognitive abilities of dogs with mucopolysaccharidosis i: Learning and memory. Animals 2020, 10, 397. [Google Scholar] [CrossRef] [Green Version]
- van der Lee, J.H.; Morton, J.; Adams, H.R.; Clarke, L.; Ebbink, B.J.; Escolar, M.L.; Giugliani, R.; Harmatz, P.; Hogan, M.; Jones, S.; et al. Cognitive endpoints for therapy development for neuronopathic mucopolysaccharidoses: Results of a consensus procedure. Mol. Genet. Metab. 2017, 121, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Janzen, D.; Delaney, K.A.; Shapiro, E.G. Cognitive and adaptive measurement endpoints for clinical trials in mucopolysaccharidoses types I, II, and III: A review of the literature. Mol. Genet. Metab. 2017, 121, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Brooks, D.A.; Evangelista, M.; Hein, L.K.; Hopwood, J.J.; Meikle, P.J. Prediction of neuropathology in mucopolysaccharidosis I patients. Mol. Genet. Metab. 2005, 84, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Langereis, E.J.; van Vlies, N.; Church, H.J.; Geskus, R.B.; Hollak, C.E.M.; Jones, S.; Kulik, W.; van Lenthe, H.; Mercer, J.; Schreider, L.; et al. Biomarker responses correlate with antibody status in mucopolysaccharidosis type I patients on long-term enzyme replacement therapy. Mol. Genet. Metab. 2015, 114, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Randall, D.R.; Sinclair, G.B.; Colobong, K.E.; Hetty, E.; Clarke, L.A. Heparin cofactor II-thrombin complex in MPS I: A biomarker of MPS disease. Mol. Genet. Metab. 2006, 88, 235–243. [Google Scholar] [CrossRef]
- Eisengart, J.B.; Pierpont, E.I.; Kaizer, A.M.; Rudser, K.D.; King, K.E.; Pasquali, M.; Polgreen, L.E.; Dickson, P.I.; Le, S.Q.; Miller, W.P.; et al. Intrathecal enzyme replacement for Hurler syndrome: Biomarker association with neurocognitive outcomes. Genet. Med. 2019, 21, 2552–2560. [Google Scholar] [CrossRef] [Green Version]
- Saville, J.T.; Thai, H.N.; Lehmann, R.J.; Derrick-Roberts, A.L.K.; Fuller, M. Subregional brain distribution of simple and complex glycosphingolipids in the mucopolysaccharidosis type I (Hurler syndrome) mouse: Impact of diet. J. Neurochem. 2017, 141, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Vedolin, L.; Schwartz, I.V.D.; Komlos, M.; Schuch, A.; Azevedo, A.C.; Vieira, T.; Maeda, F.K.; Marques Da Silva, A.M.; Giugliani, R. Brain MRI in mucopolysaccharidosis: Effect of aging and correlation with biochemical findings. Neurology 2007, 69, 917–924. [Google Scholar] [CrossRef]
- Nicolas-Jilwan, M.; AlSayed, M. Mucopolysaccharidoses: Overview of neuroimaging manifestations. Pediatr. Radiol. 2018, 48, 1503–1520. [Google Scholar] [CrossRef]
- Braunlin, E.A.; Harmatz, P.R.; Scarpa, M.; Furlanetto, B.; Kampmann, C.; Loehr, J.P.; Ponder, K.P.; Roberts, W.C.; Rosenfeld, H.M.; Giugliani, R. Cardiac disease in patients with mucopolysaccharidosis: Presentation, diagnosis and management. J. Inherit. Metab. Dis. 2011, 34, 1183–1197. [Google Scholar] [CrossRef] [Green Version]
- Wippermann, C.F.; Beck, M.; Schranz, D.; Huth, R.; Michel-Behnke, I.; Jüngst, B.K. Mitral and aortic regurgitation in 84 patients with mucopolysaccharidoses. Eur. J. Pediatr. 1995, 154, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Fesslová, V.; Corti, P.; Sersale, G.; Rovelli, A.; Russo, P.; Mannarino, S.; Butera, G.; Parini, R. The natural course and the impact of therapies of cardiac involvement in the mucopolysaccharidoses. Cardiol. Young 2009, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, D.H.; Mercer, J.; Tylee, K.; Malaiya, N.; Bonney, D.K.; Jones, S.A.; Wraith, J.E.; Wynn, R.F. Management of mucopolysaccharidosis type IH (Hurler’s syndrome) presenting in infancy with severe dilated cardiomyopathy: A single institution’s experience. J. Inherit. Metab. Dis. 2013, 36, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M.D.C.; Pennock, C.A.; Berry, P.J.; Duncan, A.W.; Cawdery, J.E.; Leonard, J.V. Hurler syndrome with cardiomyopathy in infancy. J. Pediatr. 1989, 114, 430–432. [Google Scholar] [CrossRef]
- Braunlin, E.; Miettunen, K.; Lund, T.; Luquette, M.; Orchard, P. Hematopoietic cell transplantation for severe MPS I in the first six months of life: The heart of the matter. Mol. Genet. Metab. 2019, 126, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, L.; Orchard, P.; Whitley, C.B.; Berry, J.M.; Tolar, J.; Miller, W.; Braunlin, E.A. Cardiac Ultrasound Findings in Infants with Severe (Hurler Phenotype) Untreated Mucopolysaccharidosis (MPS) Type I. Jimd. Rep. 2013, 10, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.B.; Blaser, S.I.; Burrows, P.E.; Stringer, D.A.; Clarke, J.T.R.; Thorner, P. Arteriopathy and coarctation of the abdominal aorta in children with mucopolysaccharidosis: Imaging findings. AJR Am. J. Roentgenol. 1991, 157, 819–823. [Google Scholar] [CrossRef]
- Grande-Allen, K.J.; Griffin, B.P.; Ratliff, N.B.; Cosgrove, D.M.; Vesely, I. Glycosaminoglycan profiles of myxomatous mitral leaflets and chordae parallel the severity of mechanical alterations. J. Am. Coll. Cardiol. 2003, 42, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Tseng, H.; Lawrence, B.; Grande-Allen, K. Effect of Cyclic Mechanical Strain on Glycosaminoglycan and Proteoglycan Synthesis by Heart Valve Cells. Acta Biomater. 2009, 5, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Dangel, J.H. Cardiovascular changes in children with mucopolysaccharide storage diseases and related disorders- clinical and echocardiographic findings in 64 patients. Eur. J. Pediatr. 1998, 157, 534–538. [Google Scholar] [CrossRef]
- Leal, G.N.; De Paula, A.C.; Leone, C.; Kim, C.A. Echocardiographic study of paediatric patients with mucopolysaccharidosis. Cardiol. Young 2010, 20, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.T.T.; Schaff, H.V.; Miller, F.A.; Edwards, W.D.; Karnes, P.S. Valvular heart disease in four patients with Maroteaux-Lamy syndrome. Circulation 1992, 85, 188–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, S.; Moseley, K.; Pavlova, Z. Postmortem studies on a patient with mucopolysaccharidosis type I: Histopathological findings after one year of enzyme replacement therapy. J. Inherit. Metab. Dis. 2009, 32 (Suppl. 1), S53–S57. [Google Scholar] [CrossRef]
- Braunlin, E.; Tolar, J.; Mackey-Bojack, S.; Masinde, T.; Krivit, W.; Schoen, F.J. Clear cells in the atrioventricular valves of infants with severe human mucopolysaccharidosis (Hurler syndrome) are activated valvular interstitial cells. Cardiovasc. Pathol. 2011, 20, 315–321. [Google Scholar] [CrossRef]
- Rentería, V.G.; Ferrans, V.J.; Roberts, W.C.; Renteria, V.G.; Ferrans, V.J.; Roberts, W.C. The heart in the Hurler syndrome: Gross, histologic and ultrastructural observations in five necropsy cases. Am. J. Cardiol. 1976, 38, 487–501. [Google Scholar] [CrossRef]
- Maganti, K.; Rigolin, V.H.; Sarano, M.E.; Bonow, R.O. Valvular heart disease: Diagnosis and management. Mayo Clin. Proc. 2010, 85, 483–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krovetz, L.J.; Lorincz, A.E.; Schiebler, G.L. Cardiovascular Manifestations of the Hurler Syndrome: Hemodynamic and. Circulation 1965, 31, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunlin, E.A.; Hunter, D.W.; Krivit, W.; Burke, B.A.; Hesslein, P.S.; Porter, P.T.; Whitley, C.B. Evaluation of coronary artery disease in the Hurler syndrome by angiography. Am. J. Cardiol. 1992, 69, 1487–1489. [Google Scholar] [CrossRef]
- Nemes, A.; Timmermans, R.G.M.; Wilson, J.H.P.; Soliman, O.I.I.; Krenning, B.J.; ten Cate, F.J.; Geleijnse, M.L. The mild form of mucopolysaccharidosis type I (Scheie syndrome) is associated with increased ascending aortic stiffness. Heart Vessel. 2008, 23, 108–111. [Google Scholar] [CrossRef]
- Hinek, A.; Wilson, S.E. Impaired elastogenesis in Hurler disease: Dermatan sulfate accumulation linked to deficiency in elastin-binding protein and elastic fiber assembly. Am. J. Pathol. 2000, 156, 925–938. [Google Scholar] [CrossRef]
- Boffi, L.; Russo, P.; Limongelli, G. Early diagnosis; management of cardiac manifestations in mucopolysaccharidoses: A practical guide for paediatric and adult cardiologists. Ital. J. Pediatr. 2018, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, A.M.; Dualibi, A.P.; Norato, D.; Takata, E.T.; Santos, E.S.; Valadares, E.R.; Porta, G.; de Luca, G.; Moreira, G.; Pimentel, H.; et al. Guidelines for the Management of Mucopolysaccharidosis Type I. J. Pediatr. 2009, 155. [Google Scholar] [CrossRef] [PubMed]
- Sleeper, M.M.; Kusiak, C.M.; Shofer, F.S.; O’Donnell, P.; Bryan, C.; Ponder, K.P.; Haskins, M.E. Clinical characterization of cardiovascular abnormalities associated with feline mucopolysaccharidosis I and VI. J. Inherit. Metab. Dis. 2008, 31, 424–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, M.C.; Zheng, Y.; Ryazantsev, S.; Rozengurt, N.; Roos, K.P.; Neufeld, E.F. Cardiac manifestations in the mouse model of mucopolysaccharidosis I. Mol. Genet. Metab. 2005, 86, 233–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunlin, E.; Mackey-Bojack, S.; Panoskaltsis-Mortari, A.; Berry, J.M.; McElmurry, R.T.; Riddle, M.; Sun, L.Y.; Clarke, L.A.; Tolar, J.; Blazar, B.R. Cardiac functional and histopathologic findings in humans and mice with mucopolysaccharidosis type I: Implications for assessment of therapeutic interventions in hurler syndrome. Pediatr. Res. 2006, 59, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Breider, M.A.; Shull, R.M.; Constantopoulos, G. Long-term effects of bone marrow transplantation in dogs with mucopolysaccharidosis I. Am. J. Pathol. 1989, 134, 677–692. [Google Scholar]
- Baldo, G.; Tavares, A.M.V.; Gonzalez, E.; Poletto, E.; Mayer, F.Q.; da Matte, U.S.; Giugliani, R. Progressive heart disease in mucopolysaccharidosis type I mice may be mediated by increased cathepsin B activity. Cardiovasc. Pathol. 2017, 27, 45–50. [Google Scholar] [CrossRef]
- Herati, R.; Ma, X.; Tittiger, M.; Ohlemiller, K.; Kovacs, A.; Ponder, K. Improved Retroviral Vector Design Results in Sustained Expression after Adult Gene Therapy in Mucopolysaccharidosis I Mice. J. Gene. Med. 2008, 10, 972–982. [Google Scholar] [CrossRef] [Green Version]
- Metcalf, J.A.; Ma, X.; Linders, B.; Wu, S.; Schambach, A.; Ohlemiller, K.K.; Kovacs, A.; Bigg, M.; He, L.; Tollefsen, D.M.; et al. A self-inactivating γ-retroviral vector reduces manifestations of mucopolysaccharidosis I in mice. Mol. Ther. 2010, 18, 334–342. [Google Scholar] [CrossRef] [Green Version]
- De Poswar, F.O.; de Souza, C.F.M.; Giugliani, R.; Baldo, G. Aortic root dilatation in patients with mucopolysaccharidoses and the impact of enzyme replacement therapy. Heart Vessel. 2019, 34, 290–295. [Google Scholar] [CrossRef]
- Carlisle, J.; Good, R. The Inflammatory Cycle. Am. J. Dis. Child. 1960, 99, 193–197. [Google Scholar] [CrossRef]
- Lever, R.; Page, C. Glycosaminoglycans, airways inflammation and bronchial hyperresponsiveness. Pulm. Pharmacol. Ther. 2001, 14, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.J.; Matsumoto, M.; Patel, S.; Lee, L.; Guan, J.L.; Li, S. Role of cell surface heparan sulfate proteoglycans in endothelial cell migration and mechanotransduction. J. Cell. Physiol. 2005, 203, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Adams, D.H.; Shaw, S. Proteoglycans on endothelial cells present adhesion-inducing cytokines to leukocytes. Immunol. Today 1993, 14, 111–115. [Google Scholar] [CrossRef]
- Celie, J.W.A.M.; Beelen, R.H.J.; Van Den Born, J. Heparan sulfate proteoglycans in extravasation: Assisting leukocyte guidance. Front. Biosci. 2009, 14, 4932–4949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, B.L.; Lord, M.S.; Melrose, J.; Whitelock, J.M. The Role of Heparan Sulfate in Inflammation, and the Development of Biomimetics as Anti-Inflammatory Strategies. J. Histochem. Cytochem. 2018, 66, 321–336. [Google Scholar] [CrossRef] [Green Version]
- Collins, L.E.; Troeberg, L. Heparan sulfate as a regulator of inflammation and immunity. J. Leukoc. Biol. 2019, 105, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Watson, H.A.; Holley, R.J.; Langford-Smith, K.J.; Wilkinson, F.L.; Van Kuppevelt, T.H.; Wynn, R.F.; Wraith, J.E.; Merry, C.L.R.; Bigger, B.W. Heparan sulfate inhibits hematopoietic stem and progenitor cell migration and engraftment in mucopolysaccharidosis I. J. Biol. Chem. 2014, 289, 36194–36203. [Google Scholar] [CrossRef] [Green Version]
- Johnson, G.B.; Brunn, G.J.; Kodaira, Y.; Platt, J.L. Receptor-Mediated Monitoring of Tissue Well-Being Via Detection of Soluble Heparan Sulfate by Toll-Like Receptor 4. J. Immunol. 2002, 168, 5233–5239. [Google Scholar] [CrossRef] [Green Version]
- Archer, L.D.; Langford-Smith, K.J.; Bigger, B.W.; Fildes, J.E. Mucopolysaccharide diseases: A complex interplay between neuroinflammation, microglial activation and adaptive immunity. J. Inherit. Metab. Dis. 2014, 37, 1–12. [Google Scholar] [CrossRef]
- Simonaro, C.M.; Ge, Y.; Eliyahu, E.; He, X.; Jepsen, K.J.; Schuchman, E.H. Involvement of the Toll-like receptor 4 pathway and use of TNF-α antagonists for treatment of the mucopolysaccharidoses. Proc. Natl. Acad. Sci. USA 2010, 107, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opoka-Winiarska, V.; Jurecka, A.; Emeryk, A.; Tylki-Szymanska, A. Osteoimmunology in mucopolysaccharidoses type I, II, VI and VII. Immunological regulation of the osteoarticular system in the course of metabolic inflammation. Osteroarthritis Cartil. 2013, 21, 1813–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalid, O.; Vera, M.U.; Gordts, P.L.; Ellinwood, N.M.; Schwartz, P.H.; Dickson, P.I.; Esko, J.D.; Wang, R.Y. Immune-mediated inflammation may contribute to the pathogenesis of cardiovascular disease in mucopolysaccharidosis type I. PLoS ONE 2016, 11, e0150850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, G.V.; Pasquali, M.; Polgreen, L.E.; Dickson, P.I.; Miller, W.P.; Orchard, P.J.; Lund, T.C. Elevated cerebral spinal fluid biomarkers in children with mucopolysaccharidosis I-H. Sci. Rep. 2016, 6, 4–9. [Google Scholar] [CrossRef]
- Polgreen, L.E.; Kunin-Batson, A.; Rudser, K.; Vehe, R.K.; Utz, J.J.; Whitley, C.B.; Dickson, P. Pilot study of the safety and effect of adalimumab on pain, physical function, and musculoskeletal disease in mucopolysaccharidosis types I and II. Mol. Genet. Metab. Rep. 2017, 10, 75–80. [Google Scholar] [CrossRef]
- Pereira, V.G.; Gazarini, M.L.; Rodrigues, L.C.; Da Silva, F.H.; Han, S.W.; Martins, A.M.; Tersariol, I.L.S.; D’Almeida, V. Evidence of lysosomal membrane permeabilization in mucopolysaccharidosis type I: Rupture of calcium and proton homeostasis. J. Cell. Physiol. 2010, 223, 335–342. [Google Scholar] [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.A. The mucopolysaccharidoses: A success of molecular medicine. Expert Rev. Mol. Med. 2008, 10, 1–18. [Google Scholar] [CrossRef]
- Jentsch, T.J. Chloride; the endosomal-lysosomal pathway: Emerging roles of CLC chloride transporters. J. Physiol. 2007, 578, 633–640. [Google Scholar] [CrossRef]
- Reolon, G.K.; Reinke, A.; De Oliveira, M.R.; Braga, L.M.E.; Camassola, M.; Andrades, M.É.; Moreira, J.C.F.; Nardi, N.B.; Roesler, R.; Dal-Pizzol, F. Alterations in oxidative markers in the cerebellum and peripheral organs in MPS I Mice. Cell. Mol. Neurobiol. 2009, 29, 443–448. [Google Scholar] [CrossRef]
- Reed, C.C.; Iozzo, R.V. The role of decorin in collagen fibrillogenesis and skin homeostasis. Glycoconj. J. 2002, 19, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Mouw, J.K.; Ou, G.; Weaver, V.M.; Regeneration, T. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2015, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Keene, D.R.; San Antonio, J.D.; Mayne, R.; McQuillan, D.J.; Sarris, G.; Santoro, S.A.; Iozzo, R.V. Decorin binds near the C terminus of type I collagen. J. Biol. Chem. 2000, 275, 21801–21804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maccarana, M.; Kalamajski, S.; Kongsgaard, M.; Magnusson, S.P.; Oldberg, Å.; Malmström, A. Dermatan Sulfate Epimerase 1-Deficient Mice Have Reduced Content and Changed Distribution of Iduronic Acids in Dermatan Sulfate and an Altered Collagen Structure in Skin. Mol. Cell. Biol. 2009, 29, 5517–5528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuc, I.M.; Scott, P.G. Increased diameters of collagen fibrils precipitated in vitro in the presence of decorin from various connective tissues. Connect. Tissue Res. 1997, 36, 287–296. [Google Scholar] [CrossRef]
- Young, M.F.; Bi, Y.; Ameye, L.; Chen, X.D. Biglycan knockout mice: New models for musculoskeletal diseases. Glycoconj. J. 2002, 19, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Bianco, P.; Fisher, L.W.; Longenecker, G.; Smith, E.; Goldstein, S.; Bonadio, J.; Boskey, A.; Heegaard, A.M.; Sommer, B.; et al. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat. Genet. 1998, 20, 78–82. [Google Scholar] [CrossRef]
- Chen, X.-D.; Fisher, L.W.; Robey, P.G.; Young, M.F. The small leucine-rich proteoglycan biglycan modulates BMP-4-induced osteoblast differentiation. FASEB J. 2004, 18, 948–958. [Google Scholar] [CrossRef]
- Moreno, M.; Muñoz, R.; Aroca, F.; Labarca, M.; Brandan, E.; Larraín, J. Biglycan is a new extracellular component of the Chordin-BMP4 signaling pathway. EMBO J. 2005, 24, 1397–1405. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, A.D.; Fisher, L.W.; Kilts, T.M.; Owens, R.T.; Robey, P.G.; Gutkind, J.S.; Younga, M.F. Modulation of canonical Wnt signaling by the extracellular matrix component biglycan. Proc. Natl. Acad. Sci. USA 2011, 108, 17022–17027. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Chen, S.; Goldoni, S.; Calder, B.W.; Simpson, H.C.; Owens, R.T.; McQuillan, D.J.; Young, M.F.; Iozzo, R.V.; Birk, D.E. Genetic evidence for the coordinated regulation of collagen fibrillogenesis in the cornea by decorin and biglycan. J. Biol. Chem. 2009, 284, 8888–8897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapraeger, A.; Jalkanen, M.; Bernfield, M. Cell surface proteoglycan associates with the cytoskeleton at the basolateral cell surface of mouse mammary epithelial cells. J. Cell Biol. 1986, 103, 2683–2696. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.; Bernfield, M. Cell surface proteoglycan binds mouse mammary epithelial cells to fibronectin and behaves as a receptor for interstitial matrix. J. Cell Biol. 1988, 106, 423–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef] [PubMed]
- Stepp, M.A.; Gibson, H.E.; Gala, P.H.; Drina, D.D.; Pajoohesh-Ganji, A.; Pal-Ghosh, S.; Brown, M.; Aquino, C.; Schwartz, A.M.; Goldberger, O.; et al. Defects in keratinocyte activation during wound healing in the syndecan-1-deficient mouse. J. Cell Sci. 2002, 115, 4517–4531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Y.F.; Aquino, R.S.; Park, P. Molecular functions of syndecan-1 in disease. Matrix Biol. 2012, 31, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Iozzo, R.V. Proteoglycans; neoplastic—Mesenchymal cell interactions. Hum. Pathol. 1984, 15, 2–10. [Google Scholar] [CrossRef]
- Whitelock, J.M.; Iozzo, R.V. Heparan sulfate: A complex polymer charged with biological activity. Chem. Rev. 2005, 105, 2745–2764. [Google Scholar] [CrossRef] [PubMed]
- Murdock, A.; Dodge, G.; Cohen, I.; Tuan, R.; Iozzo, R. Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan). J. Biol. Chem. 1992, 267, 8544–8557. [Google Scholar]
- Farach-Carson, M.C.; Carson, D.D. Perlecan—A multifunctional extracellular proteoglycan scaffold. Glycobiology 2007, 17, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Wilusz, R.E.; Sanchez-Adams, J.; Guilak, F. The structure and function of the pericellular matrix of articular cartilage. Matrix Biol. 2014, 39, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Ishijima, M.; Suzuki, N.; Hozumi, K.; Matsunobu, T.; Kosaki, K.; Kaneko, H.; Hassell, J.R.; Arikawa-Hirasawa, E.; Yamada, Y. Perlecan modulates VEGF signaling and is essential for vascularization in endochondral bone formation. Matrix Biol. 2012, 31, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arikawa-Hirasawa, E.; Le, A.H.; Nishino, I.; Nonaka, I.; Ho, N.C.; Francomano, C.A.; Govindraj, P.; Hassell, J.R.; Devaney, J.M.; Spranger, J.; et al. Structural and functional mutations of the perlecan gene cause Schwartz-Jampel syndrome, with myotonic myopathy and chondrodysplasia. Am. J. Hum. Genet. 2002, 70, 1368–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, D.A.; Lepori-Bui, N.; Fomin, P.V.; Sloofman, L.G.; Zhou, X.; Farach-Carson, M.C.; Wang, L.; Kirn-Safran, C.B. Deficiency in perlecan/HSPG2 during bone development enhances osteogenesis and decreases quality of adult bone in mice. Calcif. Tissue Int. 2014, 95, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Liu, Y.; Tittiger, M.; Hennig, A.; Kovacs, A.; Popelka, S.; Wang, B.; Herati, R.; Bigg, M.; Ponder, K.P. Improvements in mucopolysaccharidosis I mice after adult retroviral Vector-mediated gene therapy with immunomodulation. Mol. Ther. 2007, 15, 889–902. [Google Scholar] [CrossRef]
- Ma, X.; Tittiger, M.; Knutsen, R.H.; Kovacs, A.; Schaller, L.; Mecham, R.P.; Ponder, K.P. Upregulation of elastase proteins results in aortic dilatation in mucopolysaccharidosis I mice. Mol. Genet. Metab. 2008, 94, 298–304. [Google Scholar] [CrossRef] [Green Version]
- De Pasquale, V.; Moles, A.; Pavone, L.M. Cathepsins in the Pathophysiology of Mucopolysaccharidoses: New Perspectives for Therapy. Cells 2020, 9, 979. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, C.D.; Cipriano, S.D.; Topham, C.A.; Stevenson, D.A.; Whitehead, K.J.; Vanderhooft, S.; Presson, A.P.; McDonald, J. Localization and age distribution of telangiectases in children and adolescents with hereditary hemorrhagic telangiectasia: A retrospective cohort study. J. Am. Acad. Derm. 2019, 81, 950–955. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0–6 Months | 6–12 Months | Late |
|---|---|---|
| Recurrent rhinitis | Hearing loss | Progressive cognitive slowing, then loss |
| Upper airway obstruction | Lower airway obstruction | |
| Coarse facial features | Visual impairment | |
| Thoracolumbar kyphosis | Further musculoskeletal defects | |
| Hernias | Delayed motor milestones | |
| Hepatosplenomegaly | ||
| Cardiovascular defects | ||
| Human | Mouse | Dog | |
|---|---|---|---|
| Hearing loss | Within the first year of age | Within the first year of age | Yes |
| Middle ear | |||
| Partial obstruction of middle ear cavity | Yes | NA | Yes |
| Presence of GAG-laden cells | Yes | NA | Yes |
| Thickened tympanic membranes | Yes | NA | Yes |
| Ossicles covered by GAG-positive mucosal lining, infiltrated by GAG-laden cells | Yes | NA | Yes |
| Otitis media | Yes | Yes | No |
| Inner ear | |||
| Otitis interna | Yes | Yes | No |
| Infiltration by GAG-laden cells | Yes | Yes, but at older age | |
| Degeneration of the organ of Corti | Yes | No | |
| Loss of cochlear hair cells | Yes | Yes | No |
| Damage to cochlear nerve | Yes | Yes | |
| Damage to cochlear fibrocytes | Yes | Yes | |
| Exam | Main Targeted Area |
|---|---|
| Audiometry | Overall hearing ability |
| Bone conduction testing | Inner ear |
| Tympanometry | Middle ear |
| ART | Middle ear |
| BAEP | Inner ear |
| OAE | Inner ear (outer hair cells) |
| Static acoustic impedance | Middle ear (eardrum) |
| Human | Assessment | Animal Model | |
|---|---|---|---|
| Corneal Clouding | Yes | Slit lamp exam | Yes (all models) |
| Optic Nerve Swelling | Yes | Pupil reaction to light, visual field evaluation, fundus evaluation | |
| Retinal Degeneration | Yes (late symptom) | Color vision test | Mice and cats |
| Glaucoma | Yes (~4% of patients) | Measurement of IOP |
| Ocular Manifestation | Test |
|---|---|
| Refractive errors | Visual acuity test |
| Strabismus | Stereopsis assessment |
| Corneal clouding | Slit lamp exam Iris Camera in vivo confocal microscopy |
| Glaucoma | Visual field exam IOP Optical coherence tomography (OCT) |
| Retinopathy | Complete fundus exam OCT Ultrasound (A- and B-scan) Electroretinography |
| Optic nerve damage | OCT Visual field exam |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hampe, C.S.; Eisengart, J.B.; Lund, T.C.; Orchard, P.J.; Swietlicka, M.; Wesley, J.; McIvor, R.S. Mucopolysaccharidosis Type I: A Review of the Natural History and Molecular Pathology. Cells 2020, 9, 1838. https://doi.org/10.3390/cells9081838
Hampe CS, Eisengart JB, Lund TC, Orchard PJ, Swietlicka M, Wesley J, McIvor RS. Mucopolysaccharidosis Type I: A Review of the Natural History and Molecular Pathology. Cells. 2020; 9(8):1838. https://doi.org/10.3390/cells9081838
Chicago/Turabian StyleHampe, Christiane S., Julie B. Eisengart, Troy C. Lund, Paul J. Orchard, Monika Swietlicka, Jacob Wesley, and R. Scott McIvor. 2020. "Mucopolysaccharidosis Type I: A Review of the Natural History and Molecular Pathology" Cells 9, no. 8: 1838. https://doi.org/10.3390/cells9081838