Molecular Basis for Ser/Thr Specificity in PKA Signaling

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Western Blotting
2.3. Spectrophotometric Kinase Assay
2.4. Phosphospecific Antibody-Based Kinase Assay
2.5. Radioactive Kinase Assay
2.6. Surface Plasmon Resonance (SPR)
2.7. Docking Simulations
3. Results
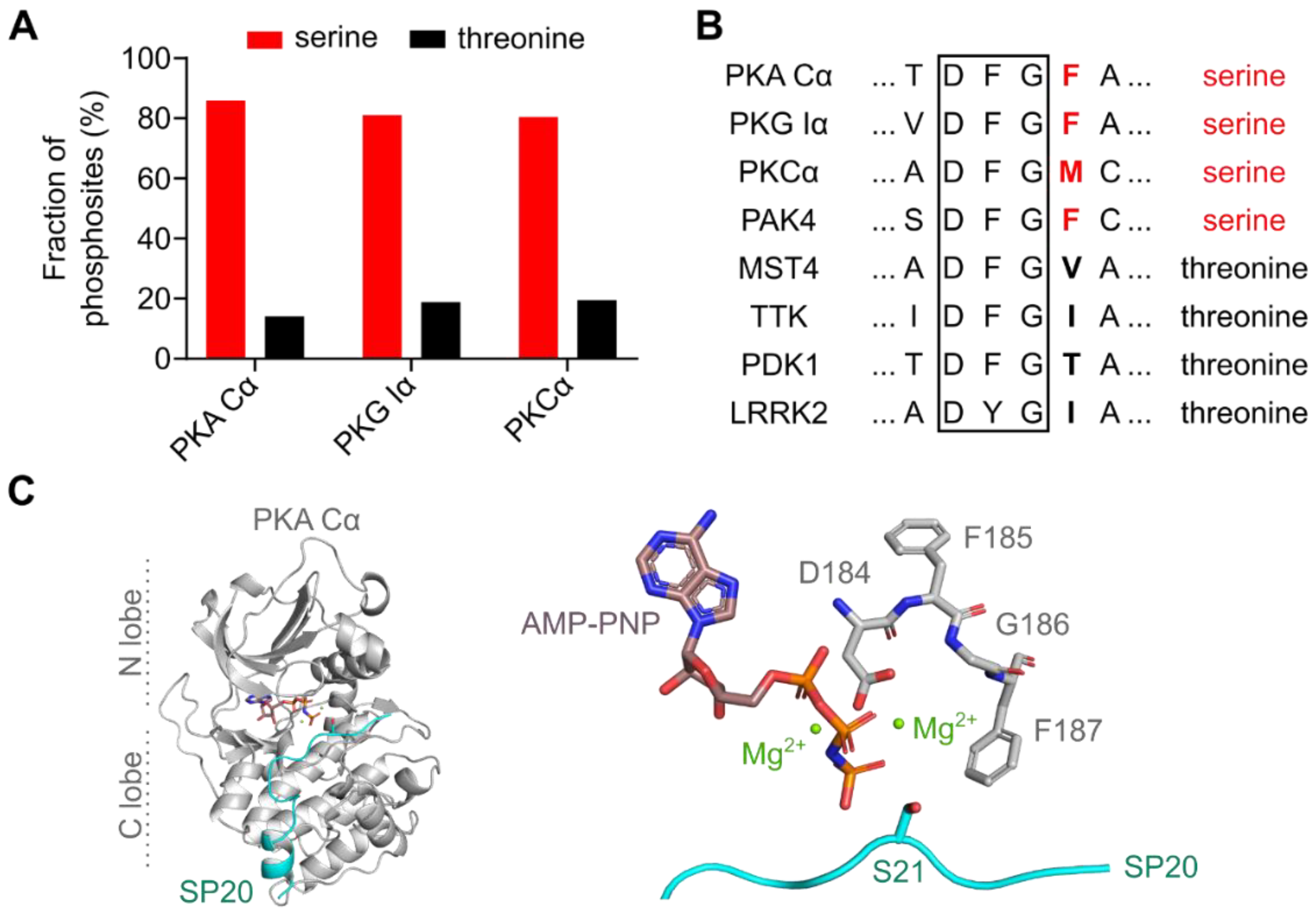
3.1. The DFG+1 Residue Determines the Serine Specificity of PKA
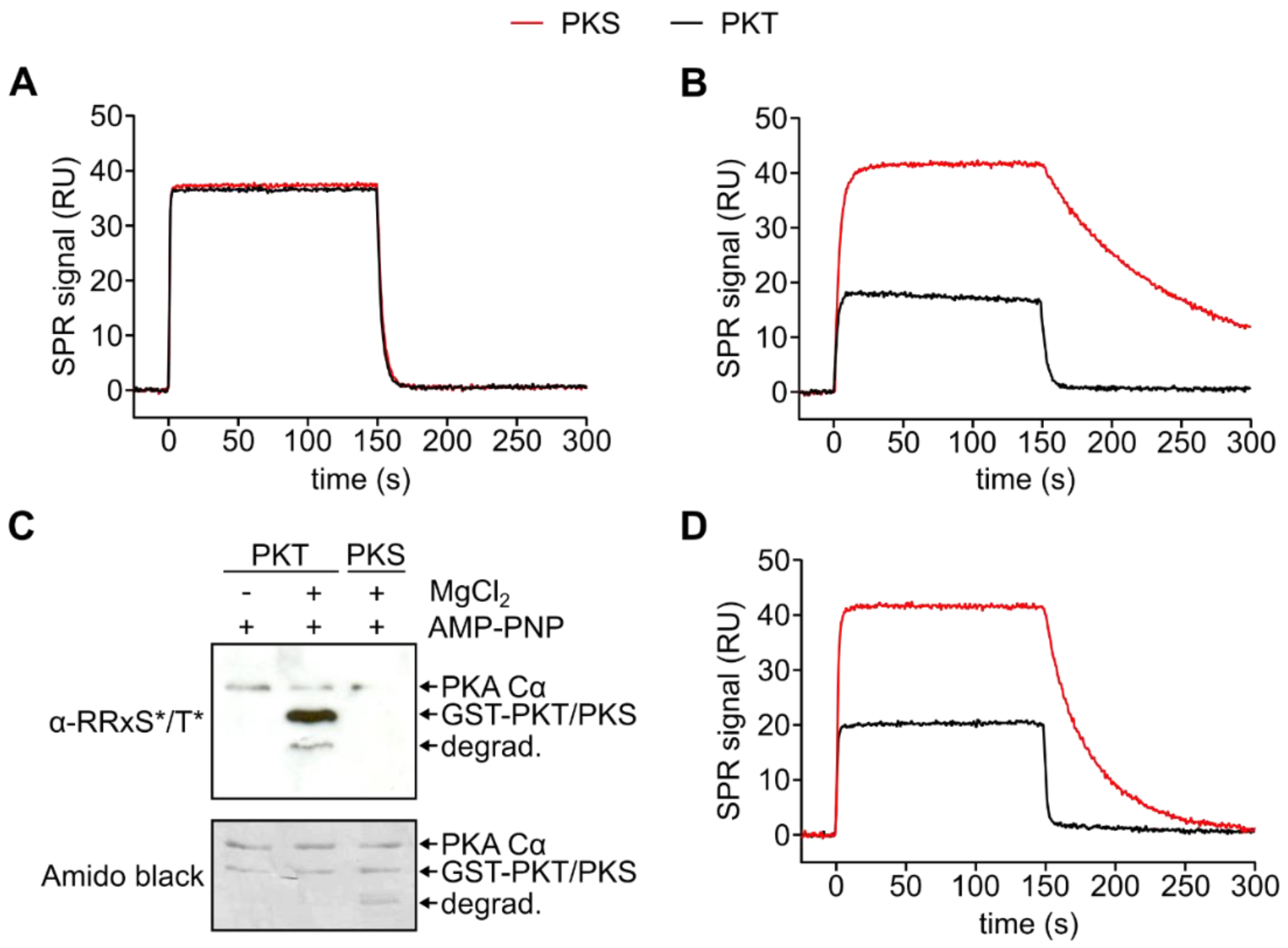
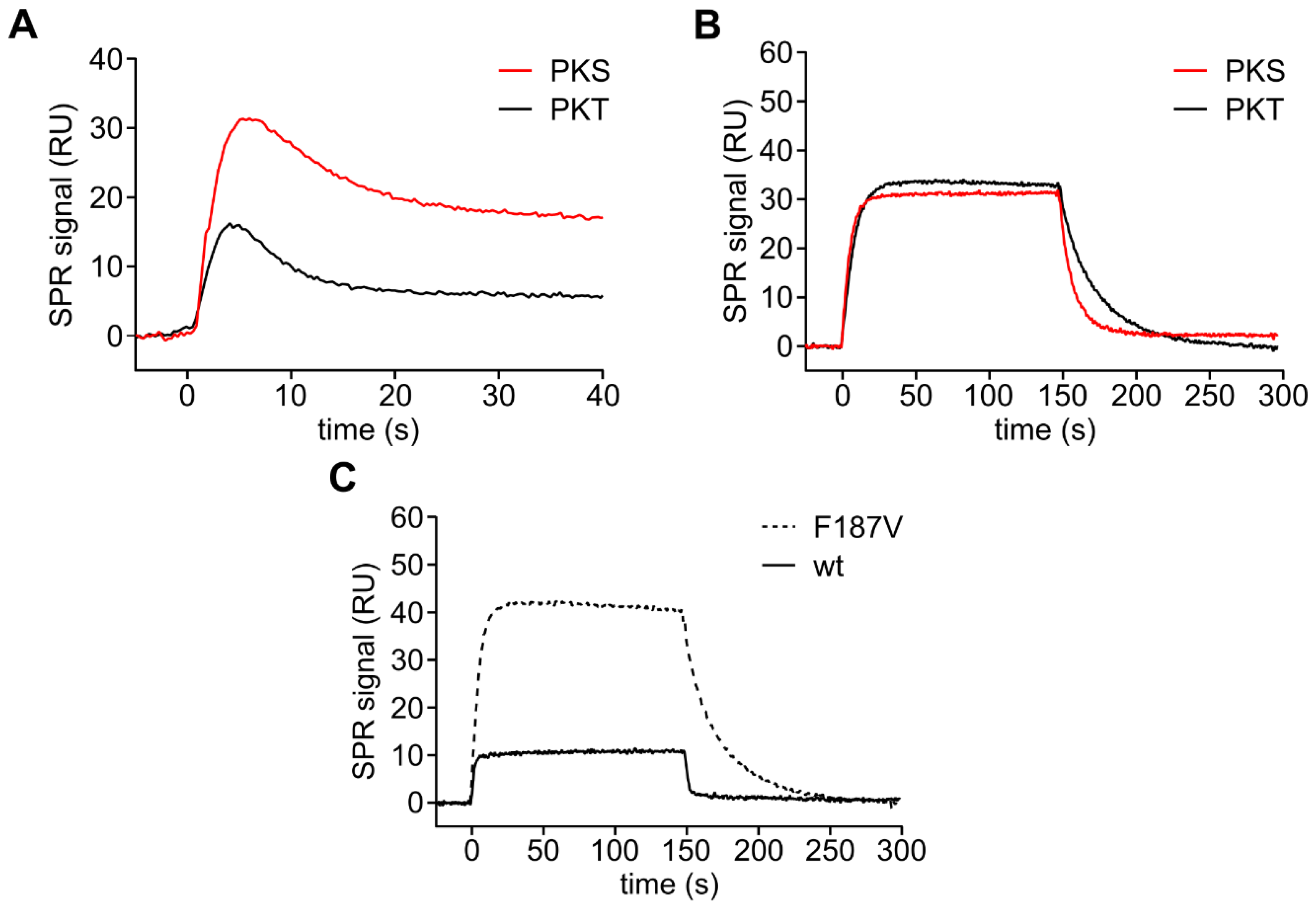
3.2. PKA Cα Phosphorylates PKT Faster than PKS
3.3. Mutation of the DFG+1 Residue Switches the Ser/Thr Specificity of PKA Cα
4. Discussion
4.1. Mutations Affecting Specificity in PKA
4.2. Mutations Affecting Specificity in Other Kinases
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research–still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, 2012–2015. [Google Scholar] [CrossRef] [PubMed]
- Schernthaner-Reiter, M.H.; Trivellin, G.; Stratakis, C.A. Chaperones, somatotroph tumors and the cyclic AMP (cAMP)-dependent protein kinase (PKA) pathway. Mol. Cell. Endocrinol. 2020, 499, 110607. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kim, C.; Cheng, C.Y.; Brown, S.H.J.; Wu, J.; Kannan, N. Signaling through cAMP and cAMP-dependent protein kinase: Diverse strategies for drug design. Biochim. Biophys. Acta 2008, 1784, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Cheng, C.Y.; Saldanha, S.A.; Taylor, S.S. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell 2007, 130, 1032–1043. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.A.; Ashby, C.D.; Gonzalez, C.; Calkins, D. Purification and characterization of a protein inhibitor of adenosine 3’, 5’-monophosphate-dependent protein kinases. J. Biol. Chem. 1971, 246, 1977–1985. [Google Scholar]
- Fantozzi, D.A.; Taylor, S.S.; Howard, P.W.; Maurer, R.A.; Feramisco, J.R.; Meinkoth, J.L. Effect of the thermostable protein kinase inhibitor on intracellular localization of the catalytic subunit of cAMP-dependent protein kinase. J. Biol. Chem. 1992, 267, 16824–16828. [Google Scholar]
- Kanev, G.K.; de Graaf, C.; de Esch, I.J.P.; Leurs, R.; Würdinger, T.; Westerman, B.A.; Kooistra, A.J. The Landscape of Atypical and Eukaryotic Protein Kinases. Trends Pharmacol. Sci. 2019, 40, 818–832. [Google Scholar] [CrossRef] [Green Version]
- Kannan, N.; Taylor, S.S.; Zhai, Y.; Venter, J.C.; Manning, G. Structural and functional diversity of the microbial kinome. PLoS Biol. 2007, 5, e17. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, R.A.; Quinn, A.M.; Hunter, T. Dual-specificity protein kinases: Will any hydroxyl do? Trends Biochem. Sci. 1992, 17, 114–119. [Google Scholar] [CrossRef]
- Durocher, D.; Taylor, I.A.; Sarbassova, D.; Haire, L.F.; Westcott, S.L.; Jackson, S.P.; Smerdon, S.J.; Yaffe, M.B. The molecular basis of FHA domain: Phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol. Cell 2000, 6, 1169–1182. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Müller, S.; Knapp, S. SH2 domains: Modulators of nonreceptor tyrosine kinase activity. Curr. Opin. Struct. Biol. 2009, 19, 643–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaffe, M.B.; Rittinger, K.; Volinia, S.; Caron, P.R.; Aitken, A.; Leffers, H.; Gamblin, S.J.; Smerdon, S.J.; Cantley, L.C.; Street, W. The Structural Basis for 14-3-3: Phosphopeptide Binding Specificity. Cell 1997, 91, 961–971. [Google Scholar] [CrossRef] [Green Version]
- Pinna, L.A.; Ruzzene, M. How do protein kinases recognize their substrates? Biochim. Biophys. Acta–Mol. Cell Res. 1996, 1314, 191–225. [Google Scholar] [CrossRef] [Green Version]
- Shabb, J.B. Physiological substrates of cAMP-dependent protein kinase. Chem. Rev. 2001, 101, 2381–2411. [Google Scholar] [CrossRef]
- Hennrich, M.L.; Marino, F.; Groenewold, V.; Kops, G.J.P.L.; Mohammed, S.; Heck, A.J.R. Universal quantitative kinase assay based on diagonal SCX chromatography and stable isotope dimethyl labeling provides high-definition kinase consensus motifs for PKA and human Mps1. J. Proteome Res. 2013, 12, 2214–2224. [Google Scholar] [CrossRef]
- Kennelly, P.J.; Krebs, E.G. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J. Biol. Chem. 1991, 266, 15555–15558. [Google Scholar]
- Zetterqvist, Ö.; Ragnarsson, U.; Engström, L. Substrate Specificity of Cyclic AMP-Dependent Protein Kinase. In Peptides and Protein Phosphorylation; Kemp, B.E., Ed.; CRC Press: Boca Raton, FL, USA, 1990; pp. 171–187. [Google Scholar]
- Loog, M.; Oskolkov, N.; O’Farrell, F.; Ek, P.; Järv, J. Comparison of cAMP-dependent protein kinase substrate specificity in reaction with proteins and synthetic peptides. Biochim. Biophys. Acta—Proteins Proteomics 2005, 1747, 261–266. [Google Scholar] [CrossRef]
- Kemp, B.E.; Graves, D.J.; Benjamini, E.; Krebs, E.G. Role of multiple basic residues in determining the substrate specificity of cyclic AMP-dependent protein kinase. J. Biol. Chem. 1977, 252, 4888–4894. [Google Scholar]
- Hjelmquist, G.; Andersson, J.; Edlund, B.; Engström, L. Amino acid sequence of a (32P)phosphopeptide from pig liver pyruvate kinase phosphorylated by cyclic 3′,5′-AMP-stimulated protein kinase and γ-(32P)ATP. Biochem. Biophys. Res. Commun. 1974, 61, 559–563. [Google Scholar] [CrossRef]
- Mitchell, R.D.; Glass, D.B.; Wong, C.W.; Angelos, K.L.; Walsh, D.A. Heat-stable inhibitor protein derived peptide substrate analogs: Phosphorylation by cAMP-dependent and cGMP-dependent protein kinases. Biochemistry 1995, 34, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Bastidas, A.C.; Deal, M.S.; Steichen, J.M.; Keshwani, M.M.; Guo, Y.; Taylor, S.S. Role of N-Terminal myristylation in the structure and regulation of cAMP-dependent protein kinase. J. Mol. Biol. 2012, 422, 215–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P.; Rylatt, D.B.; Nimmo, G.A. The hormonal control of glycogen metabolism: The amino acid sequence at the phosphorylation site of protein phosphatase inhibitor-1. FEBS Lett. 1977, 76, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Modi, V.; Dunbrack, R.L. Defining a new nomenclature for the structures of active and inactive kinases. Proc. Natl. Acad. Sci. USA 2019, 116, 6818–6827. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Ha, B.H.; Thévenin, A.F.; Lou, H.J.; Zhang, R.; Yip, K.Y.; Peterson, J.R.; Gerstein, M.; Kim, P.M.; Filippakopoulos, P.; et al. Identification of a Major Determinant for Serine-Threonine Kinase Phosphoacceptor Specificity. Mol. Cell 2014, 53, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Aimes, R.T.; Hemmer, W.; Taylor, S.S. Serine-53 at the tip of the glycine-rich loop of cAMP-dependent protein kinase: Role in catalysis, P-site specificity, and interaction with inhibitors. Biochemistry 2000, 39, 8325–8332. [Google Scholar] [CrossRef]
- Sims, P.C.; Moody, I.S.; Choi, Y.; Dong, C.; Iftikhar, M.; Corso, B.L.; Gul, O.T.; Collins, P.G.; Weiss, G.A. Electronic measurements of single-molecule catalysis by cAMP-dependent protein kinase A. J. Am. Chem. Soc. 2013, 135, 7861–7868. [Google Scholar] [CrossRef] [Green Version]
- Grant, B.D.; Adams, J.A. Pre-steady-state kinetic analysis of cAMP-dependent protein kinase using rapid quench flow techniques. Biochemistry 1996, 35, 2022–2029. [Google Scholar] [CrossRef]
- Zhou, J.; Adams, J.A. Participation of ADP dissociation in the rate-determining step in cAMP- dependent protein kinase. Biochemistry 1997, 36, 15733–15738. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, S.; Feramisco, J.R.; Casnellie, J.E.; Krebs, E.G.; Walsh, D.A. Studies on the kinetic mechanism of the catalytic subunit of the cAMP-dependent protein kinase. J. Biol. Chem. 1983, 258, 3693–3701. [Google Scholar] [PubMed]
- Masterson, L.R.; Cembran, A.; Shi, L.; Veglia, G. Allostery and binding cooperativity of the catalytic subunit of protein kinase a by NMR spectroscopy and molecular dynamics simulations. Adv. Protein Chem. Struct. Biol. 2012, 87, 363–389. [Google Scholar] [PubMed] [Green Version]
- Knape, M.J.; Ballez, M.; Burghardt, N.C.; Zimmermann, B.; Bertinetti, D.; Kornev, A.P.; Herberg, F.W. Divalent metal ions control activity and inhibition of protein kinases. Metallomics 2017, 9, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Knape, M.J.; Ahuja, L.G.; Bertinetti, D.; Burghardt, N.C.G.; Zimmermann, B.; Taylor, S.S.; Herberg, F.W. Divalent Metal Ions Mg2+ and Ca2+ Have Distinct Effects on Protein Kinase A Activity and Regulation. ACS Chem. Biol. 2015, 10, 2303–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, S.R.; Uhler, M.D. Affinity purification of the C alpha and C beta isoforms of the catalytic subunit of cAMP-dependent protein kinase. J. Biol. Chem. 1989, 264, 18662–18666. [Google Scholar]
- Cook, P.F.; Neville, M.E.; Vrana, K.E.; Hartl, F.T.; Roskoski, R. Adenosine cyclic 3’,5’-monophosphate dependent protein kinase: Kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry 1982, 21, 5794–5799. [Google Scholar] [CrossRef]
- Kish, V.M.; Kleinsmith, L.J. Purification and Assay of Nuclear Protein Kinases. Methods Cell Biol. 1978, 19, 101–107. [Google Scholar]
- Madhusudan; Trafny, E.A.; Xuong, N.-H.; Adams, J.A.; Eyck, L.F.T.; Taylor, S.S.; Sowadski, J.M. cAMP-dependent protein kinase: Crystallographic insights into substrate recognition and phosphotransfer. Protein Sci. 1994, 3, 176–187. [Google Scholar]
- Krieger, E.; Vriend, G. YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77, 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canutescu, A.A.; Shelenkov, A.A.; Dunbrack, R.L. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003, 12, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins Struct. Funct. Genet. 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, J.P. Stewart MOPAC: A semiempirical molecular orbital program. J. Comput. Aided. Mol. Des. 1990, 4, 1–103. [Google Scholar]
- Klamt, A. Conductor-like screening model for real solvents: A new approach to the quantitative calculation of solvation phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 56531, 1157–1174. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Roberts, B.P.; Chakravorty, D.K.; Merz, K.M. Rational design of particle mesh ewald compatible lennard-jones parameters for +2 metal cations in explicit solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastidas, A.C.; Deal, M.S.; Steichen, J.M.; Guo, Y.; Wu, J.; Taylor, S.S. Phosphoryl transfer by protein kinase A is captured in a crystal lattice. J. Am. Chem. Soc. 2013, 135, 4788–4798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, B.; Schweinsberg, S.; Drewianka, S.; Herberg, F.W. Effect of metal ions on high-affinity binding of pseudosubstrate inhibitors to PKA. Biochem. J. 2008, 413, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beuschlein, F.; Fassnacht, M.; Assié, G.; Calebiro, D.; Stratakis, C.A.; Osswald, A.; Ronchi, C.L.; Wieland, T.; Sbiera, S.; Faucz, F.R.; et al. Constitutive activation of PKA catalytic subunit in adrenal cushing’s syndrome. N. Engl. J. Med. 2014, 370, 1019–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espiard, S.; Knape, M.J.; Bathon, K.; Assié, G.; Rizk-Rabin, M.; Faillot, S.; Luscap-Rondof, W.; Abid, D.; Guignat, L.; Calebiro, D.; et al. Activating PRKACB somatic mutation in cortisol-producing adenomas. JCI Insight 2018, 3, e98296. [Google Scholar] [CrossRef] [Green Version]
- Knighton, D.R.; Zheng, J.; Ten Eyck, L.F.; Ashford, V.A.; Xuong, N.H.; Taylor, S.S.; Sowadski, J.M. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 1991, 253, 407–414. [Google Scholar] [CrossRef]
- Zetterqvist, Ö.; Ragnarsson, U.; Humble, E.; Berglund, L.; Engström, L. The minimum substrate of cyclic AMP-stimulated protein kinase, as studied by synthetic peptides representing the phosphorylatable site of pyruvate kinase (type L) of rat liver. Biochem. Biophys. Res. Commun. 1976, 70, 696–703. [Google Scholar] [CrossRef]
- Herberg, F.W.; Taylor, S.S. Physiological inhibitors of the catalytic subunit of cAMP-dependent protein kinase: Effect of MgATP on protein-protein interactions. Biochemistry 1993, 32, 14015–14022. [Google Scholar] [CrossRef]
- Johnson, D.A.; Akamine, P.; Radzio-Andzelm, E.; Madhusudan, M.; Taylor, S.S. Dynamics of cAMP-dependent protein kinase. Chem. Rev. 2001, 101, 2243–2270. [Google Scholar] [CrossRef]
- Chessa, G.; Borin, G.; Marchiori, F.; Meggio, F.; Brunati, A.M.; Pinna, L.A. Synthetic peptides reproducing the site phosphorylated by cAMP-dependent protein kinase in protein phosphatase inhibitor-1: Effect of structural modifications on the phosphorylation efficiency. Eur. J. Biochem. 1983, 135, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Kemp, B.E.; Parker, M.W.; Hu, S.; Tiganis, T.; House, C. Substrate and pseudosubstrate interactions with protein kinases: Determinants of specificity. Trends Biochem. Sci. 1994, 19, 440–444. [Google Scholar] [CrossRef]
- Zhang, W.; Han, Q.; Liu, Z.; Zheou, W.; Cao, Q.; Zhou, W. Exome sequencing reveals a de novo PRKG1 mutation in a sporadic patient with aortic dissection. BMC Med. Genet. 2018, 19, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, C.; Wang, Y.; Olivieri, C.; Karamafrooz, A.; Casby, J.; Bathon, K.; Calebiro, D.; Gao, J.; Bernlohr, D.A.; Taylor, S.S.; et al. Cushing’s syndrome driver mutation disrupts protein kinase A allosteric network, altering both regulation and substrate specificity. Sci. Adv. 2019, 5, 1–12. [Google Scholar] [CrossRef]
- Lubner, J.M.; Dodge-Kafka, K.L.; Carlson, C.R.; Church, G.M.; Chou, M.F.; Schwartz, D. Cushing’s syndrome mutant PKA L205R exhibits altered substrate specificity. FEBS Lett. 2017, 591, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Bathon, K.; Weigand, I.; Vanselow, J.T.; Ronchi, C.L.; Sbiera, S.; Schlosser, A.; Fassnacht, M.; Calebiro, D. Alterations in protein kinase a substrate specificity as a potential cause of cushing syndrome. Endocrinology 2019, 160, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.A.; Pacold, M.E.; Cervantes, C.L.; Lim, D.; Lou, H.J.; Ottina, K.; Gray, N.S.; Turk, B.E.; Yaffe, M.B.; Sabatini, D.M. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 2013, 341. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, J.; Oliver, C.; Shenolikar, S.; Brautigan, D.L. Mutations of the serine phosphorylated in the protein phosphatase-1-binding motif in the skeletal muscle glycogen-targeting subunit. Biochem. J. 2000, 346, 77–82. [Google Scholar] [CrossRef]
- Dent, P.; Campbell, D.G.; Hubbard, M.J.; Cohen, P. Multisite phosphorylation of the glycogen-binding subunit of protein phosphatase-1G by cyclic AMP-dependent protein kinase and glycogen synthase kinase-3. FEBS Lett. 1989, 248, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Martin, I.; Kim, J.W.; Dawson, V.L.; Dawson, T.M. LRRK2 pathobiology in Parkinson’s disease. J. Neurochem. 2014, 131, 554–565. [Google Scholar] [CrossRef] [Green Version]
- Nichols, R.J.; Dzamko, N.; Hutt, J.E.; Cantley, L.C.; Deak, M.; Moran, J.; Bamborough, P.; Reith, A.D.; Alessi, D.R. Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson’s disease. Biochem. J. 2009, 424, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, S.; Bender, S.; Kang, S.; Lin, R.; Glicksman, M.A.; Liu, M. The Parkinson disease-linked LRRK2 protein mutation I2020T stabilizes an active state conformation leading to increased kinase activity. J. Biol. Chem. 2014, 289, 13042–13053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreadi, C.; Cheung, L.K.; Giblett, S.; Patel, B.; Jin, H.; Mercer, K.; Kamata, T.; Lee, P.; Williams, A.; McMahon, M.; et al. The intermediate-activity L597V BRAF mutant acts as an epistatic modifier of oncogenic RAS by enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev. 2012, 26, 1945–1958. [Google Scholar] [CrossRef] [Green Version]
- Howard, C.J.; Hanson-Smith, V.; Kennedy, K.J.; Miller, C.J.; Lou, H.J.; Johnson, A.D.; Turk, B.E.; Holt, L.J. Ancestral resurrection reveals evolutionary mechanisms of kinase plasticity. Elife 2014, 3, 1–22. [Google Scholar] [CrossRef]
- Bremmer, S.C.; Hall, H.; Martinez, J.S.; Eissler, C.L.; Hinrichsen, T.H.; Rossie, S.; Parker, L.L.; Hall, M.C.; Charbonneau, H. Cdc14 phosphatases preferentially dephosphorylate a subset of cyclin-dependent kinase (Cdk) sites containing phosphoserine. J. Biol. Chem. 2012, 287, 1662–1669. [Google Scholar] [CrossRef] [Green Version]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PKA Cα | Substrate | kcat (s−1) | KM (µM) | kcat/KM (×105 M−1s−1) |
|---|---|---|---|---|
| Wt | S-Kemptide | 19.8 ± 1.0 | 19.8 ± 3.9 | 10.3 ± 1.7 |
| T-Kemptide | 7.5 ± 0.8 | 861 ± 184 | 0.09 ± 0.02 | |
| F187V | S-Kemptide | 35.7 ± 0.7 | 81.3 ± 27.1 | 4.8 ± 1.6 |
| T-Kemptide | 39.2 ± 3.7 | 31.5 ± 16.0 | 11.3 ± 4.0 | |
| F187I | S-Kemptide | 25.4 ± 5.8 | 92.2 ± 3.8 | 2.8 ± 0.8 |
| T-Kemptide | 43.0 ± 4.7 | 19.8 ± 3.3 | 22.1 ± 4.5 | |
| F187T | S-Kemptide | 15.7 ± 0.3 | 23.0 ± 3.1 | 6.9 ± 1.2 |
| T-Kemptide | 37.1 ± 4.5 | 25.0 ± 3.4 | 14.9 ± 0.6 |
| Phosphorylated Product | ka (M−1s−1) | kd (s−1) | KD (nM) |
|---|---|---|---|
| GST-pPKS 1 | 1.7 × 106 | 0.065 | 38 |
| GST-pPKT | 1.2 × 106 | 0.41 | 342 |
| Phosphoryl Acceptor | ka (M−1s−1) | kd (s−1) | KD (nM) |
|---|---|---|---|
| GST-PKS | 2.7 × 106 | 0.89 × 10−2 | 3.3 |
| GST-PKT | 2.2 × 106 | 29 × 10−2 | 136 |
| PKA Cα | Phosphoryl Acceptor | ka (M−1s−1) | kd (s−1) | KD (nM) |
|---|---|---|---|---|
| wt | GST-PKS | 2.8 × 106 | 3.0 × 10−2 | 11 |
| GST-PKT | 1.7 × 106 | 41 × 10−2 | 241 | |
| F187V | GST-PKS | 1.1 × 106 | 7.8 × 10−2 | 71 |
| GST-PKT | 1.7 × 106 | 4.2 × 10−2 | 26 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knape, M.J.; Wallbott, M.; Burghardt, N.C.G.; Bertinetti, D.; Hornung, J.; Schmidt, S.H.; Lorenz, R.; Herberg, F.W. Molecular Basis for Ser/Thr Specificity in PKA Signaling. Cells 2020, 9, 1548. https://doi.org/10.3390/cells9061548
Knape MJ, Wallbott M, Burghardt NCG, Bertinetti D, Hornung J, Schmidt SH, Lorenz R, Herberg FW. Molecular Basis for Ser/Thr Specificity in PKA Signaling. Cells. 2020; 9(6):1548. https://doi.org/10.3390/cells9061548
Chicago/Turabian StyleKnape, Matthias J., Maximilian Wallbott, Nicole C. G. Burghardt, Daniela Bertinetti, Jan Hornung, Sven H. Schmidt, Robin Lorenz, and Friedrich W. Herberg. 2020. "Molecular Basis for Ser/Thr Specificity in PKA Signaling" Cells 9, no. 6: 1548. https://doi.org/10.3390/cells9061548