IL7-IL12 Engineered Mesenchymal Stem Cells (MSCs) Improve A CAR T Cell Attack Against Colorectal Cancer Cells

 and
and 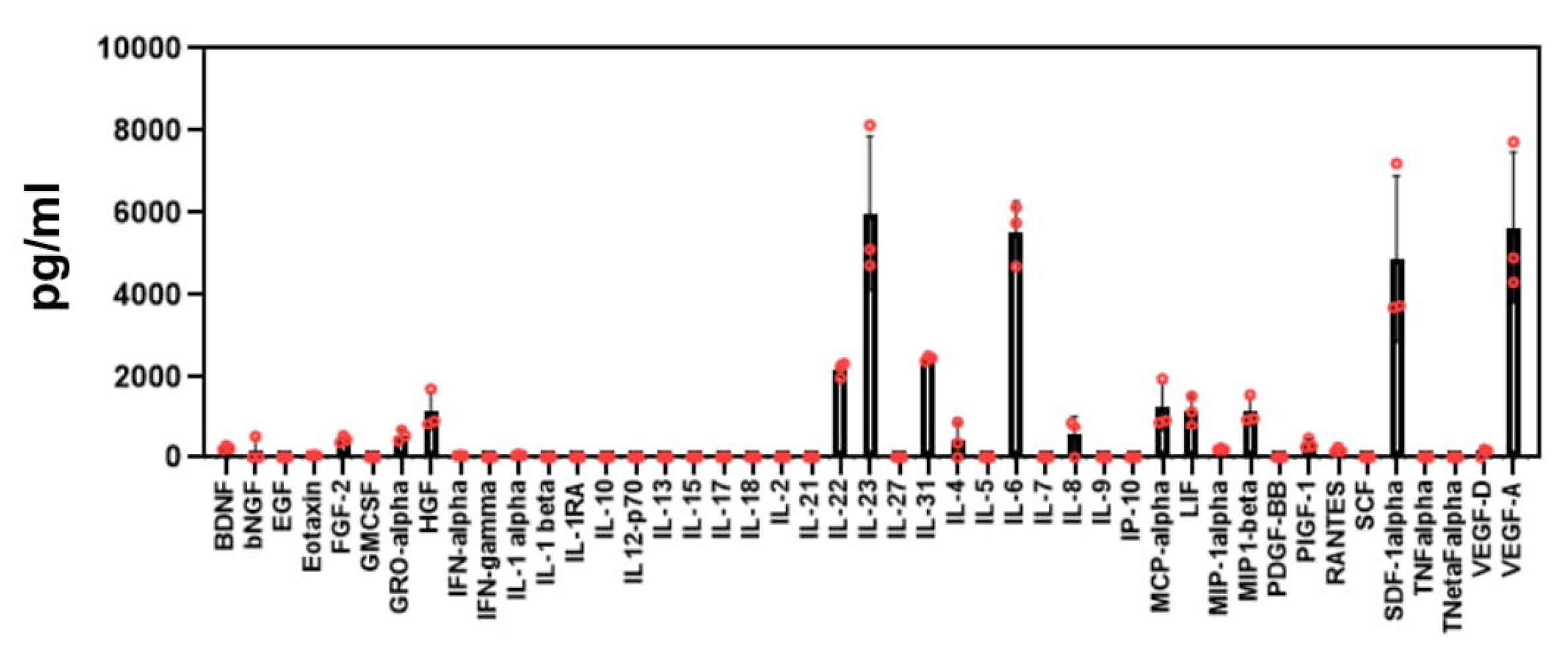{kind=link}
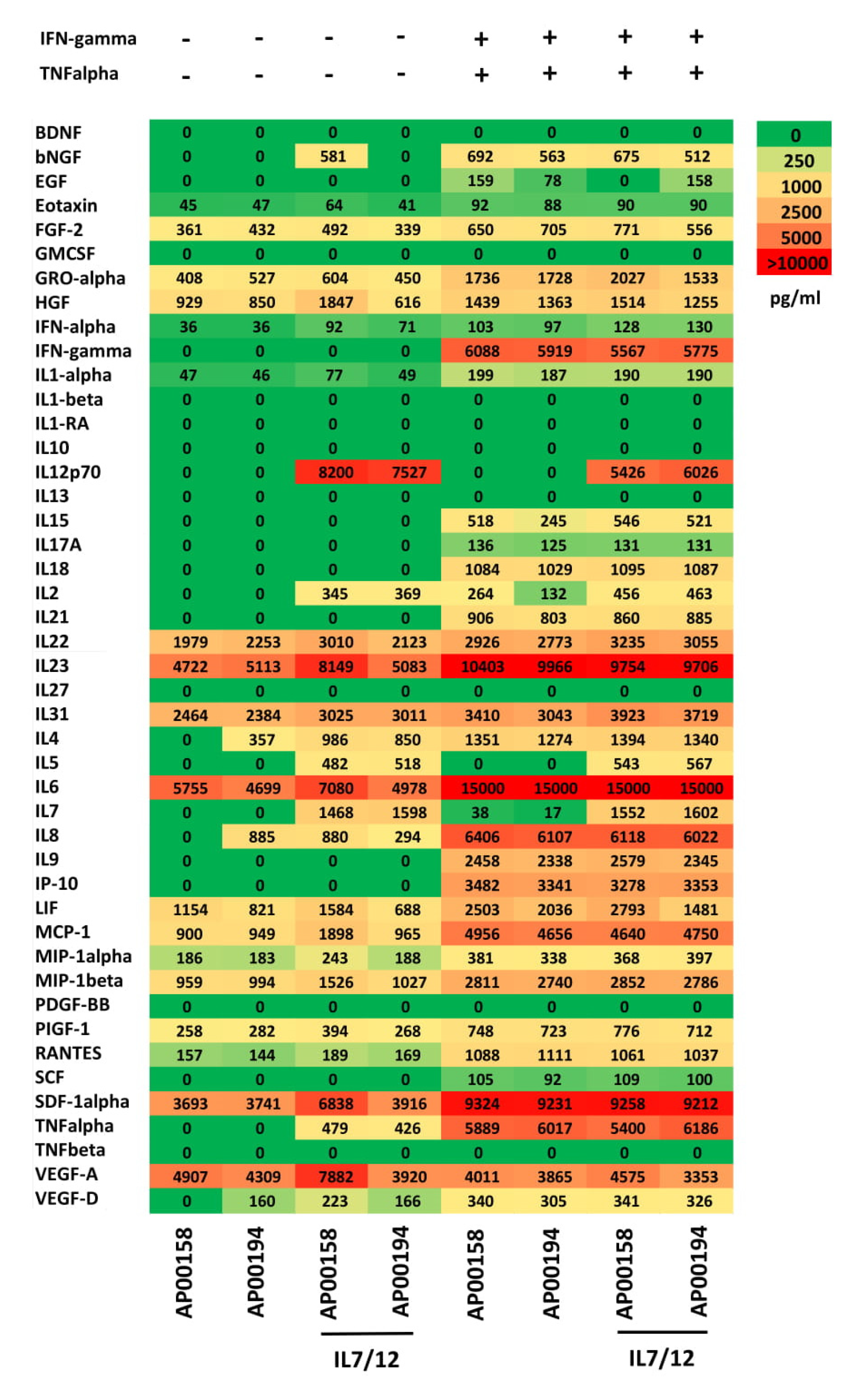{kind=link}
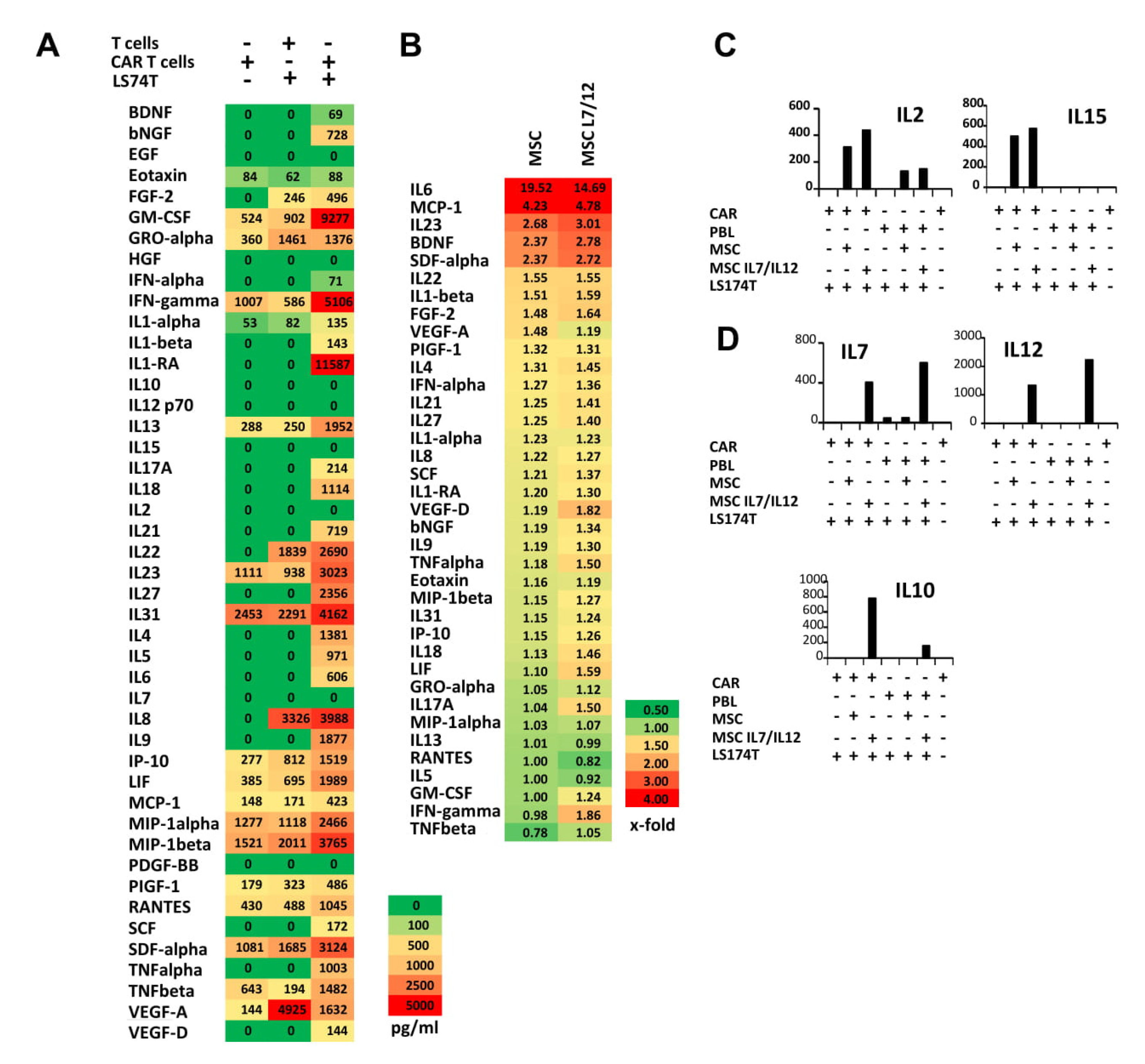{kind=link}
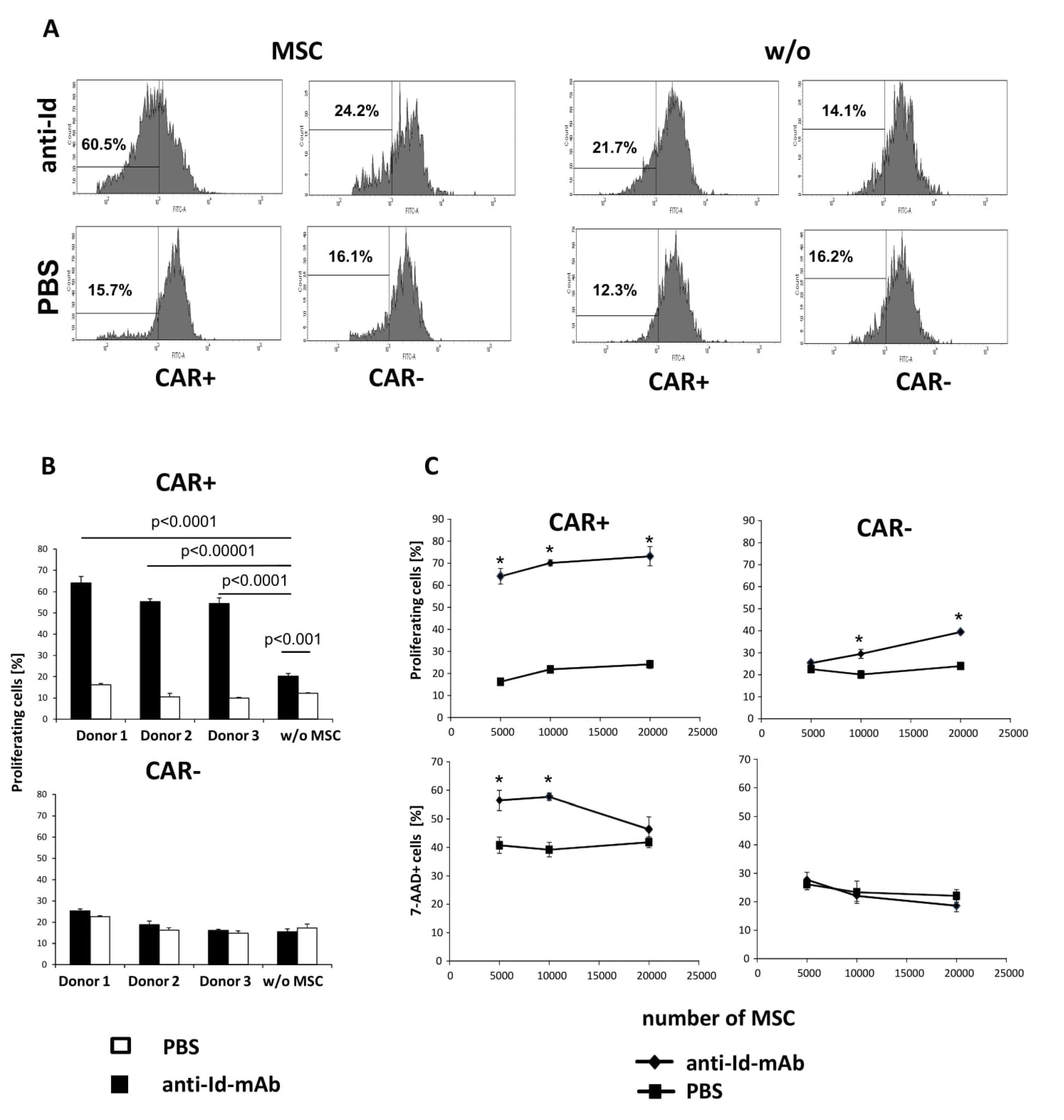{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell lines and Reagents
2.2. Preparation of Human T Cells
2.3. Isolation and Generation of Human Mesenchymal Stem Cells (MSC)
2.4. Generation of γ-Retroviral Vector to Engineer MSC
2.5. Chimeric Antigen Receptors (CARs)
2.6. T Cell Modification
2.7. Flow Cytometry
2.8. Activation of CAR T Cells
2.9. ELISA
2.10. Multiplex Immunoassay
2.11. CAR T Cell Mediated Suppression of Tumor Growth
2.12. Statistics
3. Results
3.1. CAR T Cells Modulate the Activity of MSCs
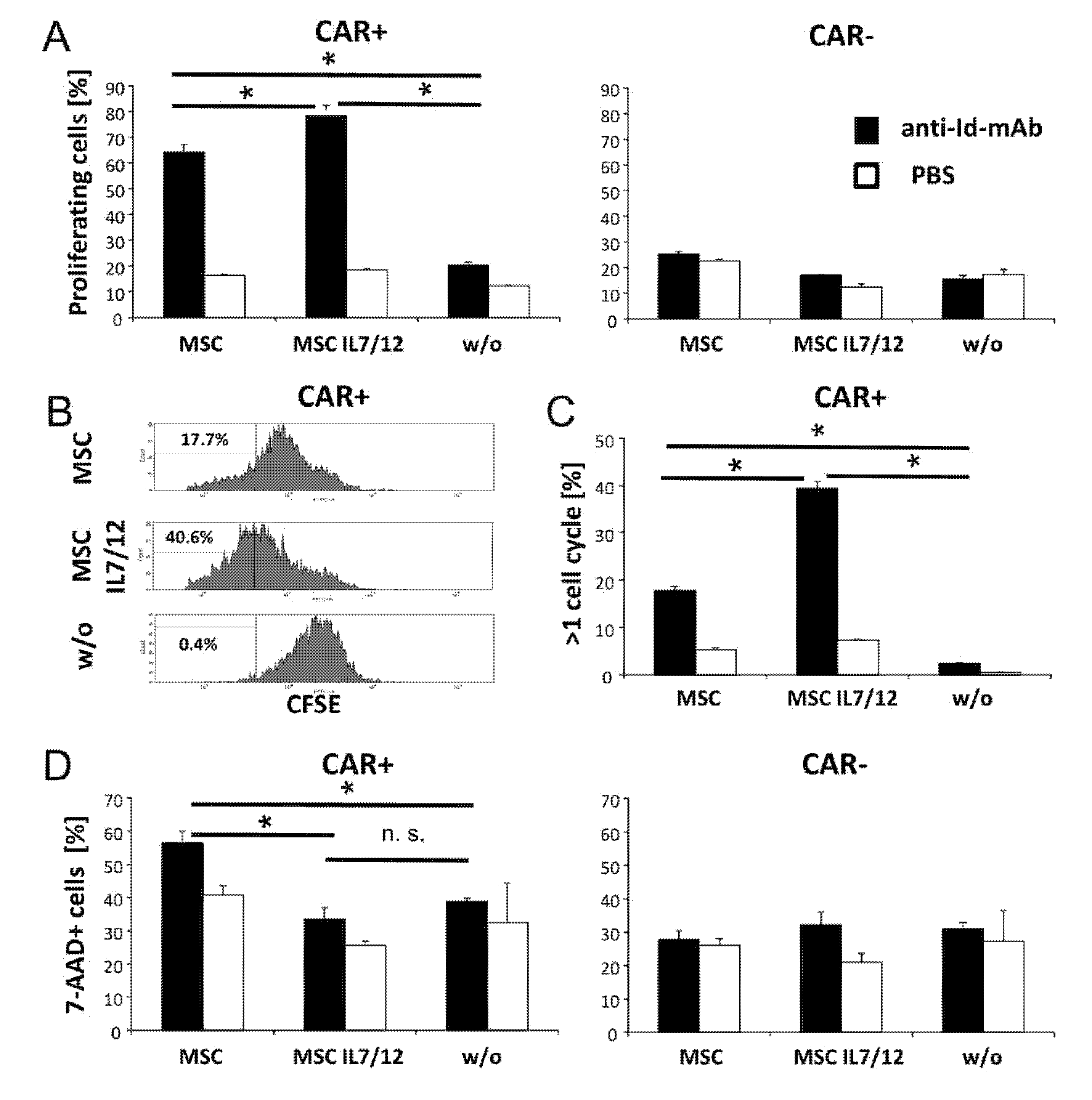
3.2. MSCs Improved Antigen-Specific Amplification and Survival of CAR T Cells
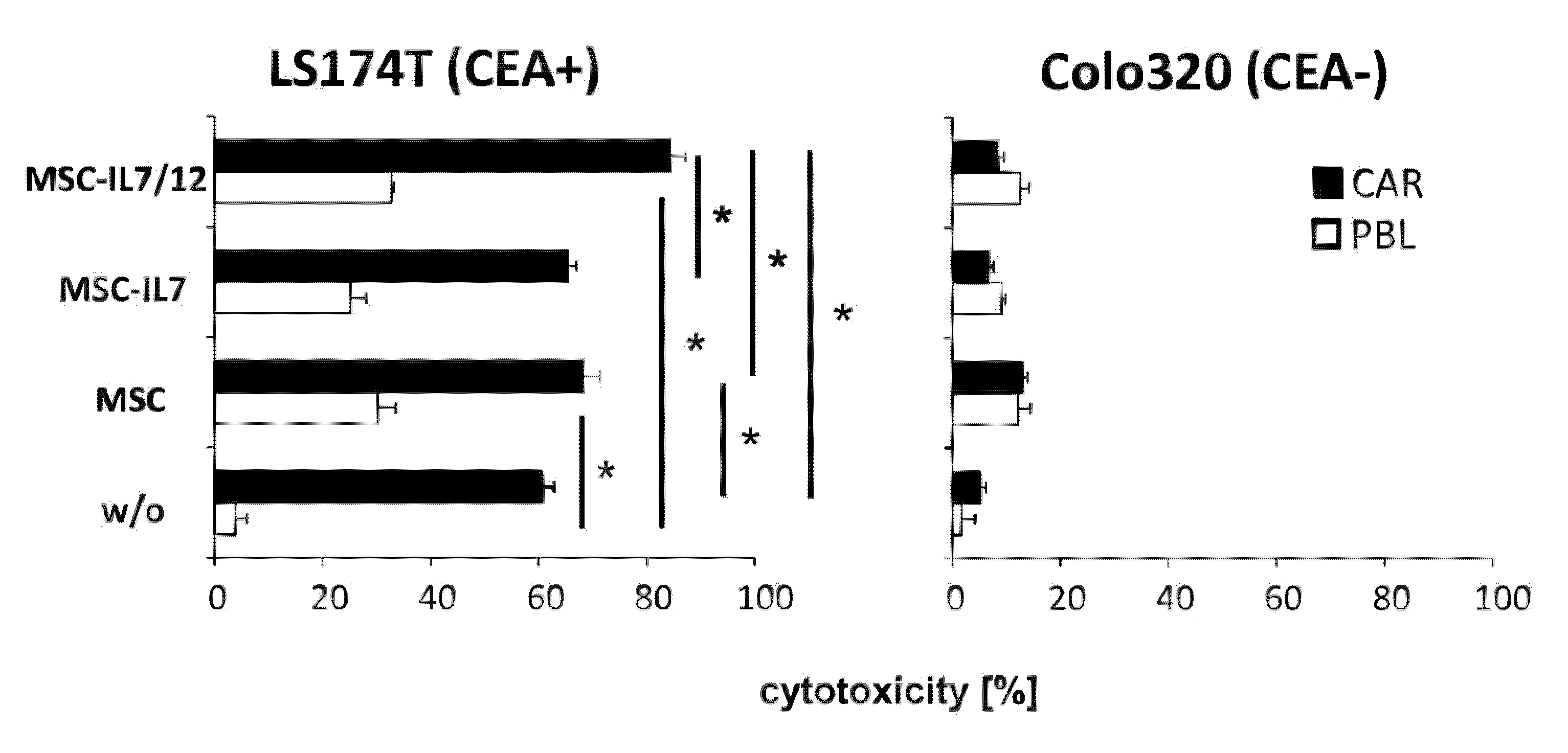
3.3. IL7 and IL12 Engineered MSCs Modulate the Cytotoxic CAR T Cell Attack
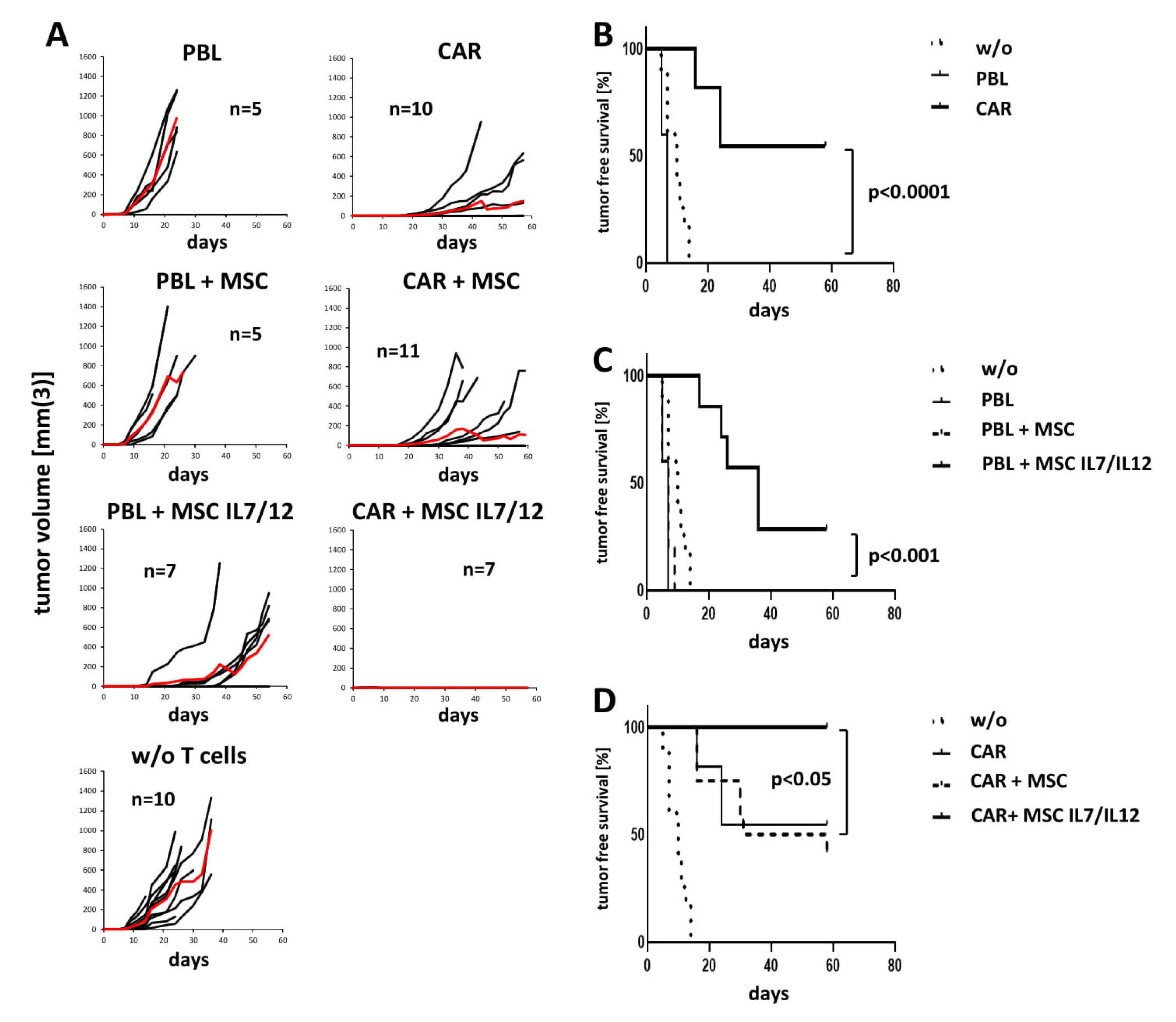
3.4. IL7 and IL12 Secreting MSCs Sustain the Overall Anti-Tumor Response in a Transplant Tumor Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, R.H. T cell anergy. Annu. Rev. Immunol. 2003, 21, 305–334. [Google Scholar] [CrossRef]
- Melzer, C.; Yang, Y.; Hass, R. Interaction of MSC with tumor cells. Cell Commun. Signal 2016, 14, 20. [Google Scholar] [CrossRef] [Green Version]
- Almalki, S.G.; Agrawal, D.K. Key transcription factors in the differentiation of mesenchymal stem cells. Differentiation 2016, 92, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Zimmerlin, L.; Park, T.S.; Zambidis, E.T.; Donnenberg, V.S.; Donnenberg, A.D. Mesenchymal stem cell secretome and regenerative therapy after cancer. Biochimie 2013, 95, 2235–2245. [Google Scholar] [CrossRef]
- Poggi, A.; Varesano, S.; Zocchi, M.R. How to Hit Mesenchymal Stromal Cells and Make the Tumor Microenvironment Immunostimulant Rather Than Immunosuppressive. Front. Immunol. 2018, 9, 262. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.E.; Ayoub, N.; Agrawal, D.K. Mesenchymal stem cells and cutaneous wound healing: Novel methods to increase cell delivery and therapeutic efficacy. Stem Cell Res. Ther. 2016, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Huang, S.; He, N.; Liu, C.; Chen, Y.; Liu, Y.; Mi, X.; Li, N.; Sun, P.; Li, Z.; et al. Inflammatory Human Umbilical Cord-Derived Mesenchymal Stem Cells Promote Stem Cell-Like Characteristics of Cancer Cells in an IL-1β-Dependent Manner. Biomed. Res. Int. 2018, 2018, 7096707. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. Exosome and mesenchymal stem cell cross-talk in the tumor microenvironment. Semin. Immunol. 2018, 35, 69–79. [Google Scholar] [CrossRef]
- Ryan, J.M.; Barry, F.P.; Murphy, J.M.; Mahon, B.P. Mesenchymal stem cells avoid allogeneic rejection. J. Inflamm. (Lond.) 2005, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Kidd, S.; Spaeth, E.; Dembinski, J.L.; Dietrich, M.; Watson, K.; Klopp, A.; Battula, V.L.; Weil, M.; Andreeff, M.; Marini, F.C. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 2009, 27, 2614–2623. [Google Scholar] [CrossRef] [Green Version]
- Studeny, M.; Marini, F.C.; Champlin, R.E.; Zompetta, C.; Fidler, I.J.; Andreeff, M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002, 62, 3603–3608. [Google Scholar]
- Studeny, M.; Marini, F.C.; Dembinski, J.L.; Zompetta, C.; Cabreira-Hansen, M.; Bekele, B.N.; Champlin, R.E.; Andreeff, M. Mesenchymal stem cells: Potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J. Natl. Cancer Inst. 2004, 96, 1593–1603. [Google Scholar] [CrossRef] [Green Version]
- Loebinger, M.R.; Eddaoudi, A.; Davies, D.; Janes, S.M. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer Res. 2009, 69, 4134–4142. [Google Scholar] [CrossRef] [Green Version]
- Attar, R.; Sajjad, F.; Qureshi, M.Z.; Tahir, F.; Hussain, E.; Fayyaz, S.; Farooqi, A.A. TRAIL based therapy: Overview of mesenchymal stem cell based delivery and miRNA controlled expression of TRAIL. Asian Pac. J. Cancer Prev. 2014, 15, 6495–6497. [Google Scholar] [CrossRef] [Green Version]
- Menon, L.G.; Shi, V.J.; Carroll, R.S. Mesenchymal stromal cells as a drug delivery system. In StemBook; Harvard Stem Cell Institute: Cambridge, MA, USA, 2008. [Google Scholar]
- Relation, T.; Yi, T.; Guess, A.J.; La Perle, K.; Otsuru, S.; Hasgur, S.; Dominici, M.; Breuer, C.; Horwitz, E.M. Intratumoral Delivery of Interferonγ-Secreting Mesenchymal Stromal Cells Repolarizes Tumor-Associated Macrophages and Suppresses Neuroblastoma Proliferation In Vivo. Stem Cells 2018, 36, 915–924. [Google Scholar] [CrossRef] [Green Version]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef]
- Hombach, A.; Heuser, C.; Gerken, M.; Fischer, B.; Lewalter, K.; Diehl, V.; Pohl, C.; Abken, H. T cell activation by recombinant FcepsilonRI gamma-chain immune receptors: An extracellular spacer domain impairs antigen-dependent T cell activation but not antigen recognition. Gene Ther. 2000, 7, 1067–1075. [Google Scholar] [CrossRef] [Green Version]
- Suerth, J.D.; Maetzig, T.; Galla, M.; Baum, C.; Schambach, A. Self-inactivating alpharetroviral vectors with a split-packaging design. J. Virol. 2010, 84, 6626–6635. [Google Scholar] [CrossRef] [Green Version]
- Hermann, F.G.; Egerer, L.; Brauer, F.; Gerum, C.; Schwalbe, H.; Dietrich, U.; von Laer, D. Mutations in gp120 contribute to the resistance of human immunodeficiency virus type 1 to membrane-anchored C-peptide maC46. J. Virol. 2009, 83, 4844–4853. [Google Scholar] [CrossRef] [Green Version]
- Hombach, A.; Hombach, A.A.; Abken, H. Adoptive immunotherapy with genetically engineered T cells: Modification of the IgG1 Fc “spacer” domain in the extracellular moiety of chimeric antigen receptors avoids “off-target” activation and unintended initiation of an innate immune response. Gene Ther. 2010, 17, 1206–1213. [Google Scholar] [CrossRef] [Green Version]
- Weijtens, M.E.; Willemsen, R.A.; Hart, E.H.; Bolhuis, R.L. A retroviral vector system “STITCH” in combination with an optimized single chain antibody chimeric receptor gene structure allows efficient gene transduction and expression in human T lymphocytes. Gene Ther. 1998, 5, 1195–1203. [Google Scholar] [CrossRef] [Green Version]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J. Immunol 2001, 167, 6123–6131. [Google Scholar] [CrossRef] [Green Version]
- Golumba-Nagy, V.; Kuehle, J.; Abken, H. Genetic Modification of T Cells with Chimeric Antigen Receptors: A Laboratory Manual. Hum. Gene Ther. Methods 2017, 28, 302–309. [Google Scholar] [CrossRef]
- Hombach, A.A.; Görgens, A.; Chmielewski, M.; Murke, F.; Kimpel, J.; Giebel, B.; Abken, H. Superior Therapeutic Index in Lymphoma Therapy: CD30(+) CD34(+) Hematopoietic Stem Cells Resist a Chimeric Antigen Receptor T-cell Attack. Mol. Ther. 2016, 24, 1423–1434. [Google Scholar] [CrossRef] [Green Version]
- Jost, L.M.; Kirkwood, J.M.; Whiteside, T.L. Improved short- and long-term XTT-based colorimetric cellular cytotoxicity assay for melanoma and other tumor cells. J. Immunol. Methods 1992, 147, 153–165. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Kofler, D.M.; Chmielewski, M.; Rappl, G.; Hombach, A.; Riet, T.; Schmidt, A.; Hombach, A.A.; Wendtner, C.-M.; Abken, H. CD28 Costimulation Impairs the Efficacy of a Redirected T-cell Antitumor Attack in the Presence of Regulatory T cells Which Can Be Overcome by Preventing Lck Activation. Mol. Ther. 2011, 19, 760–767. [Google Scholar] [CrossRef]
- Garcia, C.A.; Wang, H.; Benakanakere, M.R.; Barrett, E.; Kinane, D.F.; Martin, M. c-jun controls the ability of IL-12 to induce IL-10 production from human memory CD4+ T cells. J. Immunol. 2009, 183, 4475–4482. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Tazetdinova, L.G.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Application of Mesenchymal Stem Cells for Therapeutic Agent Delivery in Anti-tumor Treatment. Front. Pharmacol. 2018, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- de Witte, S.F.H.; Merino, A.M.; Franquesa, M.; Strini, T.; van Zoggel, J.A.A.; Korevaar, S.S.; Luk, F.; Gargesha, M.; O’Flynn, L.; Roy, D.; et al. Cytokine treatment optimises the immunotherapeutic effects of umbilical cord-derived MSC for treatment of inflammatory liver disease. Stem Cell Res. Ther. 2017, 8, 140. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Cheng, J.; Shi, P.; Huang, J. [Human umbilical cord mesenchymal stem cells with adenovirus-mediated interleukin 12 gene transduction inhibits the growth of ovarian carcinoma cells both in vitro and in vivo]. Nan Fang Yi Ke Da Xue Xue Bao 2011, 31, 903–907. [Google Scholar] [PubMed]
- Nakamura, K.; Ito, Y.; Kawano, Y.; Kurozumi, K.; Kobune, M.; Tsuda, H.; Bizen, A.; Honmou, O.; Niitsu, Y.; Hamada, H. Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Ther. 2004, 11, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhao, J.; Xu, J.; Wen, Y. Mesenchymal stem cells genetically modified by lentivirus-mediated interleukin-12 inhibit malignant ascites in mice. Exp. Ther. Med. 2014, 8, 1330–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohme, M.; Maire, C.L.; Geumann, U.; Schliffke, S.; Dührsen, L.; Fita, K.D.; Akyüz, N.; Binder, M.; Westphal, M.; Guenther, C.; et al. Local intracerebral immunomodulation using interleukin-expressing mesenchymal stem cells in glioblastoma. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Perna, S.K.; Pagliara, D.; Mahendravada, A.; Liu, H.; Brenner, M.K.; Savoldo, B.; Dotti, G. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin. Cancer Res. 2014, 20, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Haabeth, O.A.W.; Lorvik, K.B.; Hammarström, C.; Donaldson, I.M.; Haraldsen, G.; Bogen, B.; Corthay, A. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat. Commun. 2011, 2, 240. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Shalapour, S.; Karin, M. Immunity, inflammation, and cancer: An eternal fight between good and evil. J. Clin. Investig. 2015, 125, 3347–3355. [Google Scholar] [CrossRef] [Green Version]
- Baglio, S.R.; Lagerweij, T.; Pérez-Lanzón, M.; Ho, X.D.; Léveillé, N.; Melo, S.A.; Cleton-Jansen, A.-M.; Jordanova, E.S.; Roncuzzi, L.; Greco, M.; et al. Blocking Tumor-Educated MSC Paracrine Activity Halts Osteosarcoma Progression. Clin. Cancer Res. 2017, 23, 3721–3733. [Google Scholar] [CrossRef] [Green Version]
- Mannino, M.H.; Zhu, Z.; Xiao, H.; Bai, Q.; Wakefield, M.R.; Fang, Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015, 367, 103–107. [Google Scholar] [CrossRef]
- Candido, J.; Hagemann, T. Cancer-related inflammation. J. Clin. Immunol. 2013, 33 (Suppl. 1), S79–S84. [Google Scholar] [CrossRef]
- Balyasnikova, I.V.; Franco-Gou, R.; Mathis, J.M.; Lesniak, M.S. Genetic modification of mesenchymal stem cells to express a single-chain antibody against EGFRvIII on the cell surface. J. Tissue Eng. Regen. Med. 2010, 4, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Golinelli, G.; Grisendi, G.; Prapa, M.; Bestagno, M.; Spano, C.; Rossignoli, F.; Bambi, F.; Sardi, I.; Cellini, M.; Horwitz, E.M.; et al. Targeting GD2-positive glioblastoma by chimeric antigen receptor empowered mesenchymal progenitors. Cancer Gene Ther. 2018. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Woo, J.S.; Jeong, C.H.; Ryu, C.H.; Lim, J.Y.; Jeun, S.-S. Effective combination therapy for malignant glioma with TRAIL-secreting mesenchymal stem cells and lipoxygenase inhibitor MK886. Cancer Res. 2012, 72, 4807–4817. [Google Scholar] [CrossRef] [Green Version]
- Sasportas, L.S.; Kasmieh, R.; Wakimoto, H.; Hingtgen, S.; van de Water, J.A.J.M.; Mohapatra, G.; Figueiredo, J.L.; Martuza, R.L.; Weissleder, R.; Shah, K. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc. Natl. Acad. Sci. USA. 2009, 106, 4822–4827. [Google Scholar] [CrossRef] [Green Version]
- Komarova, S.; Roth, J.; Alvarez, R.; Curiel, D.T.; Pereboeva, L. Targeting of mesenchymal stem cells to ovarian tumors via an artificial receptor. J. Ovarian Res. 2010, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Szoor, A.; Vaidya, A.; Velasquez, M.P.; Mei, Z.; Galvan, D.L.; Torres, D.; Gee, A.; Heczey, A.; Gottschalk, S. T Cell-Activating Mesenchymal Stem Cells as a Biotherapeutic for HCC. Mol. Ther. Oncolytics. 2017, 6, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Ziblat, A.; Nuñez, S.Y.; Raffo Iraolagoitia, X.L.; Spallanzani, R.G.; Torres, N.I.; Sierra, J.M.; Secchiari, F.; Domaica, C.I.; Fuertes, M.B.; Zwirner, N.W. Interleukin (IL)-23 Stimulates IFN-γ Secretion by CD56bright Natural Killer Cells and Enhances IL-18-Driven Dendritic Cells Activation. Front. Immunol. 2017, 8, 1959. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, D.L.; Mundhenke, T.M.; Gigley, J.P. The IL-12- and IL-23-Dependent NK Cell Response Is Essential for Protective Immunity against Secondary Toxoplasma gondii Infection. J. Immunol. 2019, 203, 2944–2958. [Google Scholar] [CrossRef]
- Xu, X.; Weiss, I.D.; Zhang, H.; Singh, S.P.; Wynn, T.A.; Wilson, M.S.; Farber, J.M. Conventional NK cells can produce IL-22 and promote host defense in K. pneumoniae pneumonia. J. Immunol. 2014, 192, 1778–1786. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hombach, A.A.; Geumann, U.; Günther, C.; Hermann, F.G.; Abken, H. IL7-IL12 Engineered Mesenchymal Stem Cells (MSCs) Improve A CAR T Cell Attack Against Colorectal Cancer Cells. Cells 2020, 9, 873. https://doi.org/10.3390/cells9040873
Hombach AA, Geumann U, Günther C, Hermann FG, Abken H. IL7-IL12 Engineered Mesenchymal Stem Cells (MSCs) Improve A CAR T Cell Attack Against Colorectal Cancer Cells. Cells. 2020; 9(4):873. https://doi.org/10.3390/cells9040873
Chicago/Turabian StyleHombach, Andreas A., Ulf Geumann, Christine Günther, Felix G. Hermann, and Hinrich Abken. 2020. "IL7-IL12 Engineered Mesenchymal Stem Cells (MSCs) Improve A CAR T Cell Attack Against Colorectal Cancer Cells" Cells 9, no. 4: 873. https://doi.org/10.3390/cells9040873