NK cells and CD38: Implication for (Immuno)Therapy in Plasma Cell Dyscrasias

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Development and Maturation of NK Cells
1.2. NK Cell Receptors
1.3. NK Cell Effector Functions
2. NK Cells and Plasma Cells Dyscrasias
3. CD38 Expression and Function in Immune (NK) Cells
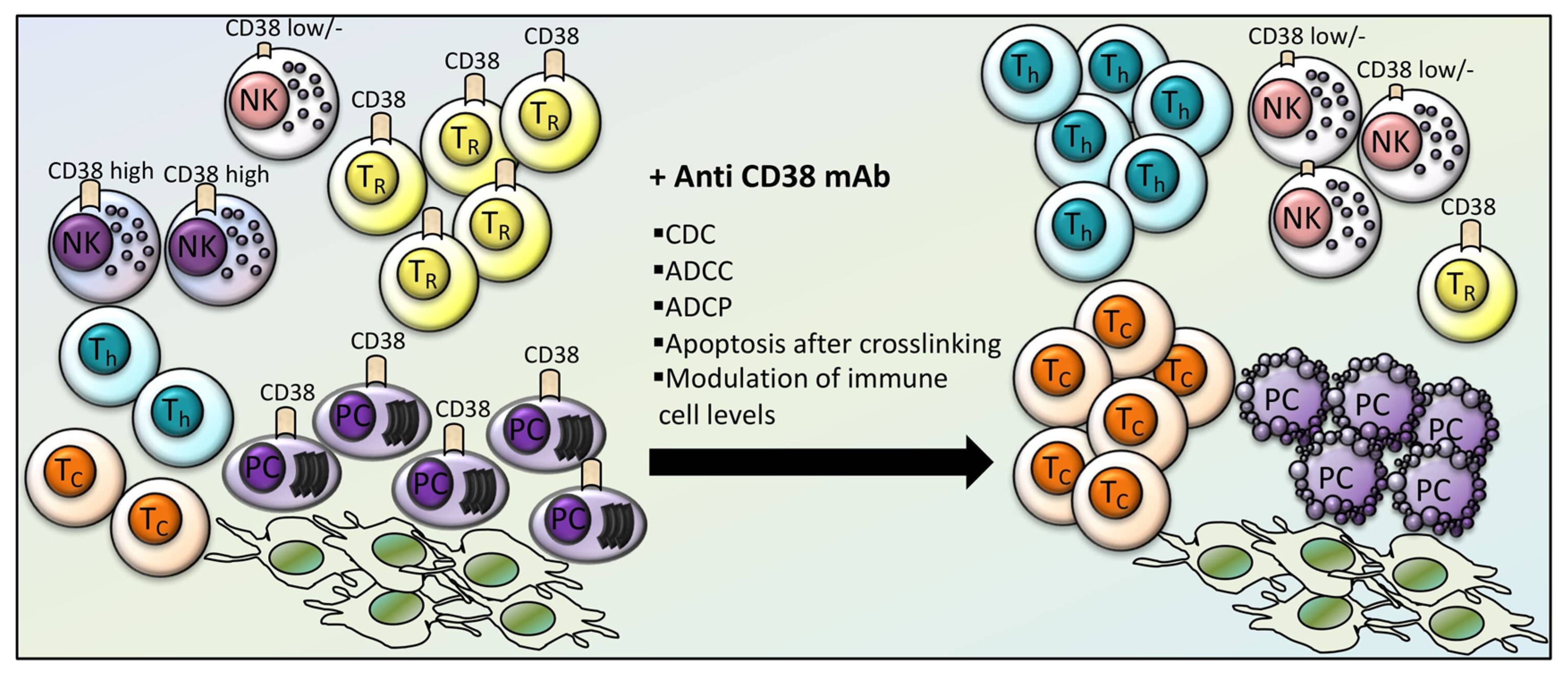
4. Effects of CD38-Directed Immunotherapy on Immune Cell Levels and Function
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrien-Elliott, M.M.; Wagner, J.A.; Fehniger, T.A. Human Cytokine-Induced Memory-Like Natural Killer Cells. J. Innate. Immun. 2015, 7, 563–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, K.S.; Hasegawa, J. Natural killer cell biology: An update and future directions. J. Allergy Clin. Immunol. 2013, 132, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.C.; Lanier, L.L. NK cell development, homeostasis and function: Parallels with CD8(+) T cells. Nat. Rev. Immunol. 2011, 11, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.C.; Theorell, J.; Entesarian, M.; Meeths, M.; Mastafa, M.; Al-Herz, W.; Frisk, P.; Gilmour, K.C.; Ifversen, M.; Langenskiold, C.; et al. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood 2013, 121, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer. Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Borrego, F.; Masilamani, M.; Marusina, A.I.; Tang, X.; Coligan, J.E. The CD94/NKG2 family of receptors: From molecules and cells to clinical relevance. Immunol. Res. 2006, 35, 263–278. [Google Scholar] [CrossRef]
- Dosani, T.; Carlsten, M.; Maric, I.; Landgren, O. The cellular immune system in myelomagenesis: NK cells and T cells in the development of MM and their uses in immunotherapies. Blood Cancer J. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- De Rossi, G.; De Sanctis, G.; Bottari, V.; Tribalto, M.; Lopez, M.; Petrucci, M.T.; Fontana, L. Surface markers and cytotoxic activities of lymphocytes in monoclonal gammopathy of undetermined significance and untreated multiple myeloma. Increased phytohemagglutinin-induced cellular cytotoxicity and inverted helper/suppressor cell ratio are features common to both diseases. Cancer Immunol. Immunother. 1987, 25, 133–136. [Google Scholar] [CrossRef] [PubMed]
- King, M.A.; Radicchi-Mastroianni, M.A. Natural killer cells and CD56+ T cells in the blood of multiple myeloma patients: Analysis by 4-colour flow cytometry. Cytometry 1996, 26, 121–124. [Google Scholar] [CrossRef]
- Omede, P.; Boccadoro, M.; Gallone, G.; Frieri, R.; Battaglio, S.; Redoglia, V.; Pileri, A. Multiple myeloma: Increased circulating lymphocytes carrying plasma cell-associated antigens as an indicator of poor survival. Blood 1990, 76, 1375–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barilà, G.; Pavan, L.; Vedovato, S.; Berno, T.; Branca, A.; Teramo, A.; Calabretto, G.; Manni, S.; Trimarco, V.; Carraro, S.; et al. Immune profiling of plasma cell dyscrasias reveals a therapy related T-cell modulation in Multiple Myeloma patients. Proceedings of 17th Internation Myeloma Workshop, Boston, MA, USA, 12–15 September 2019; p. 135. [Google Scholar]
- Pittari, G.; Vago, L.; Festuccia, M.; Bonini, C.; Mudawi, D.; Giaccone, L.; Bruno, B. Restoring Natural Killer Cell Immunity against Multiple Myeloma in the Era of New Drugs. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.M., Jr.; Cohen, A.D.; Jagannath, S.; Munshi, N.C.; Spitzer, G.; Hofmeister, C.C.; Efebera, Y.A.; Andre, P.; Zerbib, R.; Caligiuri, M.A. A Phase I Trial of the Anti-KIR Antibody IPH2101 and Lenalidomide in Patients with Relapsed/Refractory Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4055–4061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korde, N.; Carlsten, M.; Lee, M.J.; Minter, A.; Tan, E.; Kwok, M.; Manasanch, E.; Bhutani, M.; Tageja, N.; Roschewski, M.; et al. A phase II trial of pan-KIR2D blockade with IPH2101 in smoldering multiple myeloma. Haematologica 2014, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, B.; Mateos, M.V.; Sanchez-Abarca, L.I.; Puig, N.; Vidriales, M.B.; Lopez-Corral, L.; Corchete, L.A.; Hernandez, M.T.; Bargay, J.; de Arriba, F.; et al. Immune status of high-risk smoldering multiple myeloma patients and its therapeutic modulation under LenDex: A longitudinal analysis. Blood 2016, 127, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Besson, L.; Charrier, E.; Karlin, L.; Allatif, O.; Marcais, A.; Rouzaire, P.; Belmont, L.; Attal, M.; Lombard, C.; Salles, G.; et al. One-Year Follow-Up of Natural Killer Cell Activity in Multiple Myeloma Patients Treated With Adjuvant Lenalidomide Therapy. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Balasa, B.; Yun, R.; Belmar, N.A.; Fox, M.; Chao, D.T.; Robbins, M.D.; Starling, G.C.; Rice, A.G. Elotuzumab enhances natural killer cell activation and myeloma cell killing through interleukin-2 and TNF-alpha pathways. Cancer Immunol. Immunother. 2015, 64, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Lonial, S.; Vij, R.; Harousseau, J.L.; Facon, T.; Moreau, P.; Mazumder, A.; Kaufman, J.L.; Leleu, X.; Tsao, L.C.; Westland, C.; et al. Elotuzumab in combination with lenalidomide and low-dose dexamethasone in relapsed or refractory multiple myeloma. J. Clin. Oncol. 2012, 30, 1953–1959. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.; Raje, N. Anti-BCMA CAR T-cell therapy in multiple myeloma: Can we do better? Leukemia 2020, 34, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Leivas, A.; Rio, P.; Mateos, R.; Paciello, M.L.; Garcia-Ortiz, A.; Fernandez, L.; Perez-Martinez, A.; Anthony Lee, D.; Powell, D.J.; Valeri, A.; et al. NKG2D-CAR Transduced Primary Natural Killer Cells Efficiently Target Multiple Myeloma Cells. Blood 2018, 132. [Google Scholar] [CrossRef]
- Alessio, M.; Roggero, S.; Funaro, A.; De Monte, L.B.; Peruzzi, L.; Geuna, M.; Malavasi, F. CD38 molecule: Structural and biochemical analysis on human T lymphocytes, thymocytes, and plasma cells. J. Immunol. 1990, 145, 878–884. [Google Scholar]
- Jackson, D.G.; Bell, J.I. Isolation of a cDNA encoding the human CD38 (T10) molecule, a cell surface glycoprotein with an unusual discontinuous pattern of expression during lymphocyte differentiation. J. Immunol. 1990, 144, 2811–2815. [Google Scholar]
- Higuchi, Y.; Zeng, H.; Ogawa, M. CD38 expression by hematopoietic stem cells of newborn and juvenile mice. Leukemia 2003, 17, 171–174. [Google Scholar] [CrossRef] [Green Version]
- Nagler, A.; Lanier, L.L.; Cwirla, S.; Phillips, J.H. Comparative studies of human FcRIII-positive and negative natural killer cells. J. Immunol. 1989, 143, 3183–3191. [Google Scholar]
- Snoeck, H.W.; Lardon, F.; Lenjou, M.; Nys, G.; Van Bockstaele, D.R.; Peetermans, M.E. Differential regulation of the expression of CD38 and human leukocyte antigen-DR on CD34+ hematopoietic progenitor cells by interleukin-4 and interferon-gamma. Exp. Hematol. 1993, 21, 1480–1486. [Google Scholar]
- Funaro, A.; De Monte, L.B.; Dianzani, U.; Forni, M.; Malavasi, F. Human CD38 is associated to distinct molecules which mediate transmembrane signaling in different lineages. Eur. J. Immunol. 1993, 23, 2407–2411. [Google Scholar] [CrossRef]
- Kitanaka, A.; Ito, C.; Coustan-Smith, E.; Campana, D. CD38 ligation in human B cell progenitors triggers tyrosine phosphorylation of CD19 and association of CD19 with lyn and phosphatidylinositol 3-kinase. J. Immunol. 1997, 159, 184–192. [Google Scholar]
- Morra, M.; Zubiaur, M.; Terhorst, C.; Sancho, J.; Malavasi, F. CD38 is functionally dependent on the TCR/CD3 complex in human T cells. FASEB J. 1998, 12, 581–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sconocchia, G.; Titus, J.A.; Mazzoni, A.; Visintin, A.; Pericle, F.; Hicks, S.W.; Malavasi, F.; Segal, D.M. CD38 triggers cytotoxic responses in activated human natural killer cells. Blood 1999, 94, 3864–3871. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Zubiaur, M.; Gregorini, A.; Bottarel, F.; Ausiello, C.M.; Dianzani, U.; Sancho, J.; Malavasi, F. Human CD38 and CD16 are functionally dependent and physically associated in natural killer cells. Blood 2002, 99, 2490–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morandi, F.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Chiesa, S.; Imperatori, A.; Zanellato, S.; Mortara, L.; Gattorno, M.; Pistoia, V.; et al. CD56brightCD16- NK Cells Produce Adenosine through a CD38-Mediated Pathway and Act as Regulatory Cells Inhibiting Autologous CD4+ T Cell Proliferation. J. Immunol. 2015, 195, 965–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, F.K.; Bell, A.J.; Cusack, R.; Hamblin, T.J.; Slade, C.J.; Spellerberg, M.B.; Stevenson, G.T. Preliminary studies for an immunotherapeutic approach to the treatment of human myeloma using chimeric anti-CD38 antibody. Blood 1991, 77, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- de Weers, M.; Tai, Y.T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Mateos, M.V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N. Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef]
- Jansen, J.H.M.; Boross, P.; Overdijk, M.B.; van Bueren, J.J.L.; Parren, P.W.H.I.; Leusen, J.H.W. Daratumumab, a Human CD38 Antibody Induces Apoptosis of Myeloma Tumor Cells Via Fc Receptor-Mediated Crosslinking. Blood 2012, 120. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, Y.; Hughes, T.; Zhang, J.; Caligiuri, M.A.; Benson, D.M.; Yu, J. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo-Expanded Autologous NK Cells. Clin. Cancer Res. 2018, 24, 4006–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahaweni, N.M.; Bos, G.M.J.; Mitsiades, C.S.; Tilanus, M.G.J.; Wieten, L. Daratumumab augments alloreactive natural killer cell cytotoxicity towards CD38+ multiple myeloma cell lines in a biochemical context mimicking tumour microenvironment conditions. Cancer Immunol. Immunother. 2018, 67, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casneuf, T.; Xu, X.S.; Adams, H.C., 3rd; Axel, A.E.; Chiu, C.; Khan, I.; Ahmadi, T.; Yan, X.; Lonial, S.; Plesner, T.; et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood. Adv. 2017, 1, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.; Strickland, S.; Glenn, M.; Charpentier, E.; Guillemin, H.; Hsu, K.; Mikhael, J. Phase I trial of isatuximab monotherapy in the treatment of refractory multiple myeloma. Blood Cancer J. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhael, J.; Richardson, P.; Usmani, S.Z.; Raje, N.; Bensinger, W.; Karanes, C.; Campana, F.; Kanagavel, D.; Dubin, F.; Liu, Q.; et al. A phase 1b study of isatuximab plus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma. Blood 2019, 134, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Moreno, L.; Perez, C.; Zabaleta, A.; Manrique, I.; Alignani, D.; Ajona, D.; Blanco, L.; Lasa, M.; Maiso, P.; Rodriguez, I.; et al. The Mechanism of Action of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef] [Green Version]
- Moreno, L.; Zabaleta, A.; Alignani, D.; Ajona, D.; Lasa, M.; Maiso, P.; Jelinek, T.; Segura, V.; Delgado, J.A.; Rodriguez-Otero, P.; et al. Critical Analysis on the Mechanism of Action (MoA) of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma (MM). Blood 2016, 128, 2105. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, L.; Acharya, C.; An, G.; Wen, K.; Qiu, L.; Munshi, N.C.; Tai, Y.T.; Anderson, K.C. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin. Cancer Res. 2017, 23, 4290–4300. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zambello, R.; Barilà, G.; Manni, S.; Piazza, F.; Semenzato, G. NK cells and CD38: Implication for (Immuno)Therapy in Plasma Cell Dyscrasias. Cells 2020, 9, 768. https://doi.org/10.3390/cells9030768
Zambello R, Barilà G, Manni S, Piazza F, Semenzato G. NK cells and CD38: Implication for (Immuno)Therapy in Plasma Cell Dyscrasias. Cells. 2020; 9(3):768. https://doi.org/10.3390/cells9030768
Chicago/Turabian StyleZambello, Renato, Gregorio Barilà, Sabrina Manni, Francesco Piazza, and Gianpietro Semenzato. 2020. "NK cells and CD38: Implication for (Immuno)Therapy in Plasma Cell Dyscrasias" Cells 9, no. 3: 768. https://doi.org/10.3390/cells9030768