Motor Neuron Generation from iPSCs from Identical Twins Discordant for Amyotrophic Lateral Sclerosis

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Sample Collection
2.2. Induced Pluripotent Stem Cells (iPSC) Reprogramming and Validation
2.3. Whole Genome Sequencing
2.4. Stem Cell Culture
2.5. Motor Neuron Differentiation
2.6. Immunocytochemistry
2.7. Protein Isolation and Analysis
2.8. Statistical Analysis
3. Results
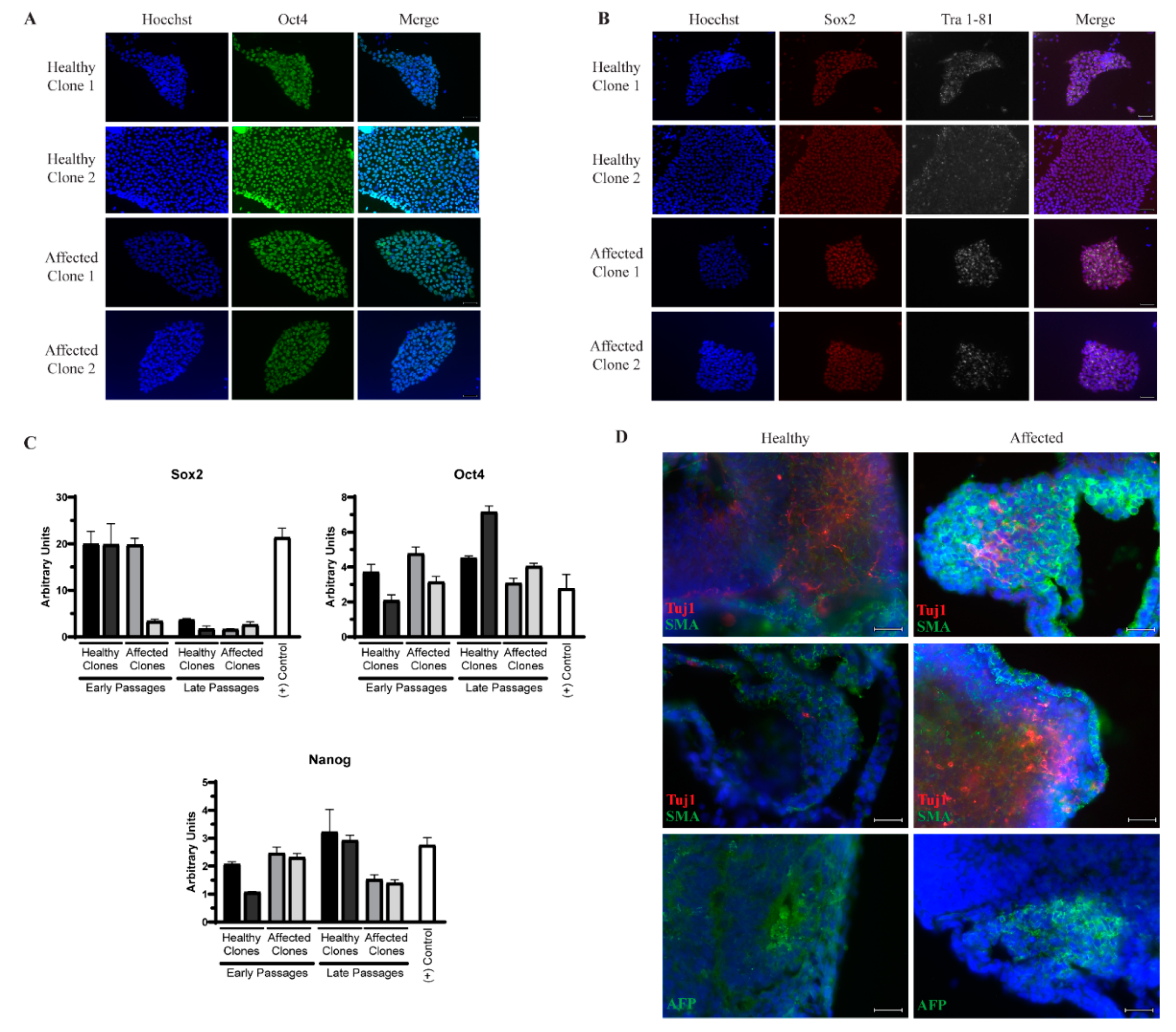
3.1. Generation of Induced Pluripotent Stem Cells (iPSCs) from Identical Twins Discordant for Amyotrophic Lateral Sclerosis (ALS)
3.2. Whole Genome Sequencing (WGS) Reveals no Meaningful Genetic Differences
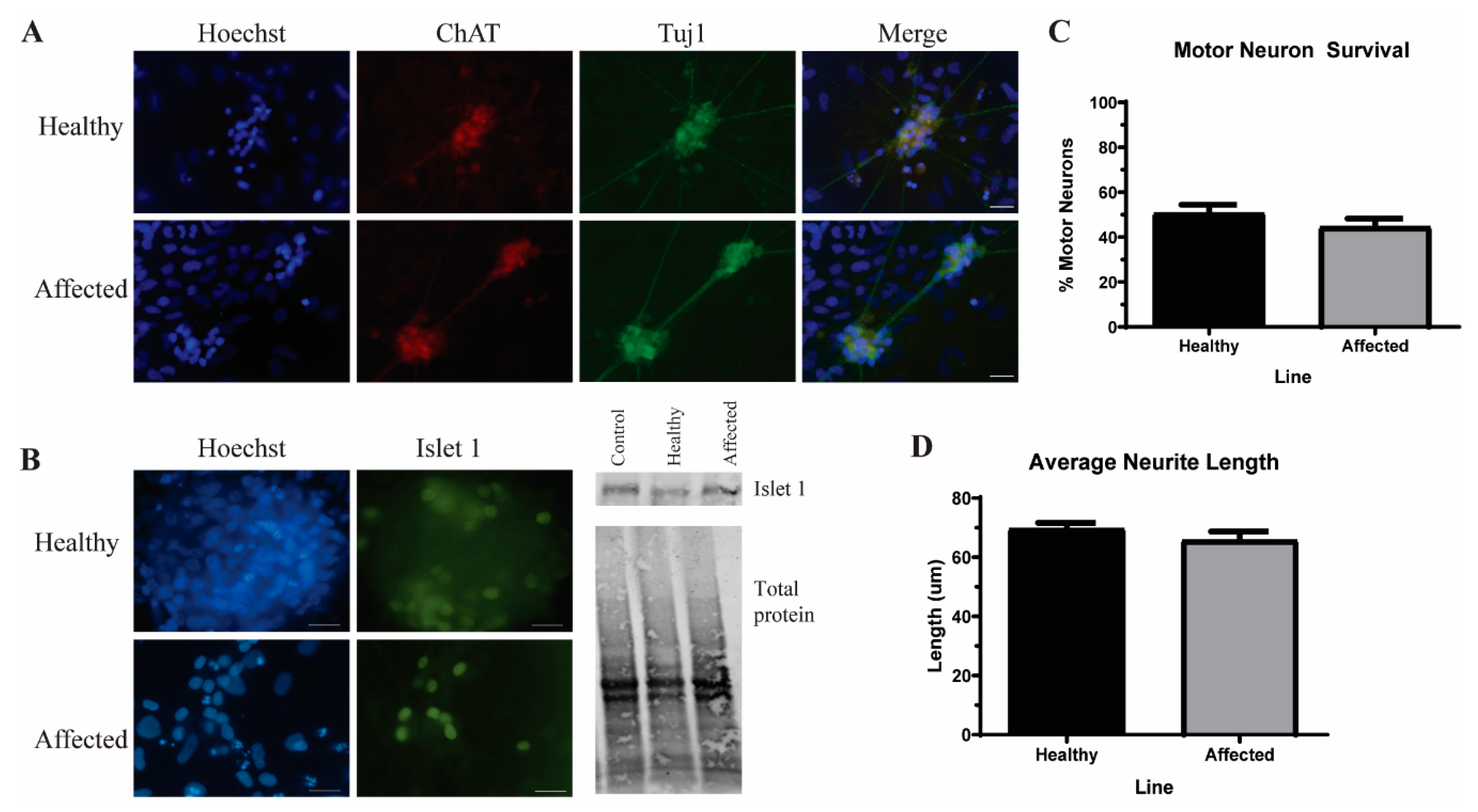
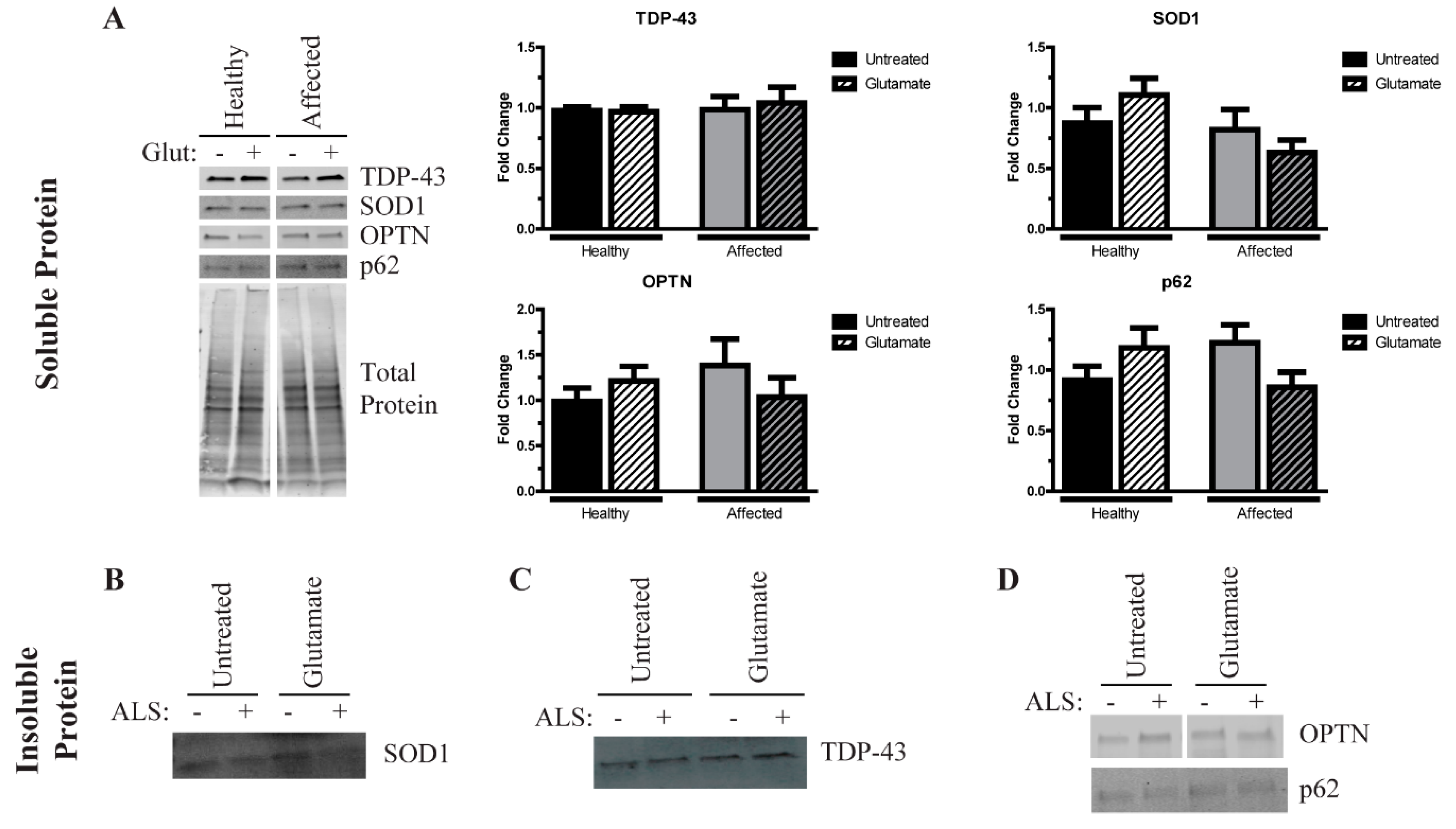
3.3. Induced Pluripotent Stem Cells (iPSC)-Derived Motor Neurons (MNs) Have Similar Levels of Insoluble Proteins while Maintaining Viability
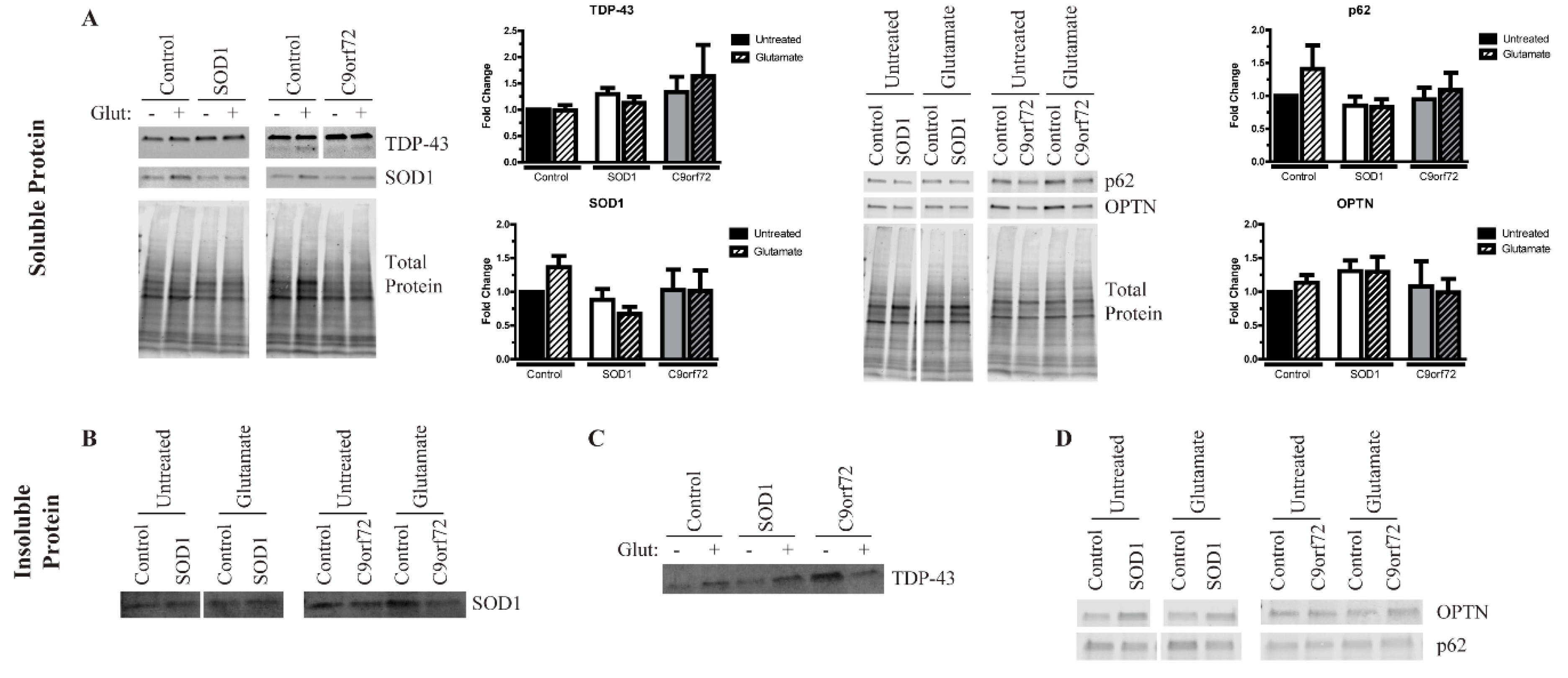
3.4. Excess Glutamate Increases the Insolubility of Aggregation-Prone Proteins
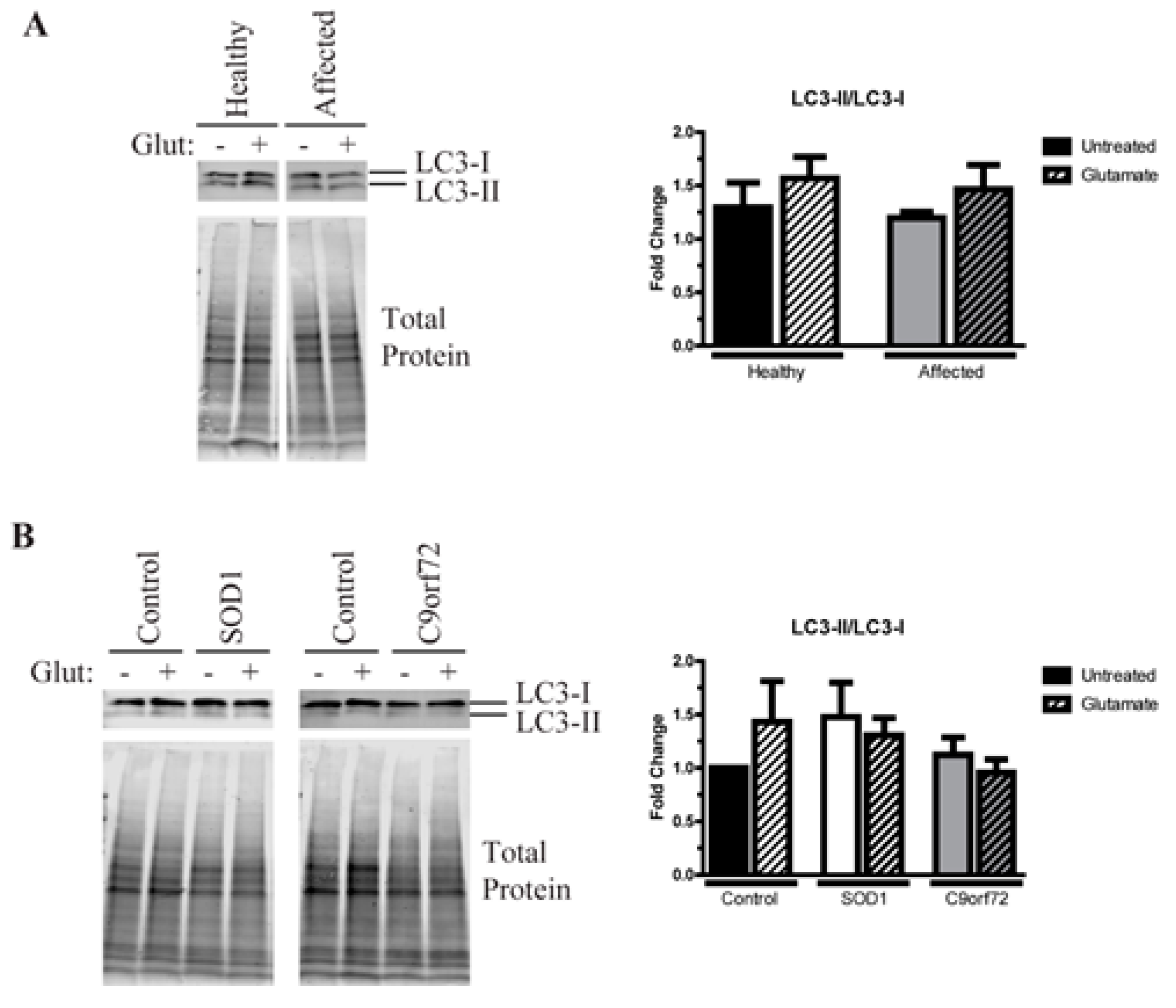
3.5. Alterations in Autophagic Flux in Amyotrophic Lateral Sclerosis (ALS) Motor Neurons (MNs)
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Zufiria, M.; Gil-Bea, F.J.; Fernandez-Torron, R.; Poza, J.J.; Munoz-Blanco, J.L.; Rojas-Garcia, R.; Riancho, J.; de Munain, A.L. ALS: A bucket of genes, environment, metabolism and unknown ingredients. Prog. Neurobiol. 2016, 142, 104–129. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; DeFranco, D.B.; Orr, H.T.; Zoghbi, H.Y. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998, 19, 148–154. [Google Scholar] [CrossRef]
- Ii, K.; Ito, H.; Tanaka, K.; Hirano, A. Immunocytochemical co-localization of the proteasome in ubiquitinated structures in neurodegenerative diseases and the elderly. J. Neuropathol. Exp. Neurol. 1997, 56, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Behl, C.; Davis, J.B.; Lesley, R.; Schubert, D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 1994, 77, 817–827. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, B.; Faure, A.J.; Seuma, M.; Schmiedel, J.M.; Tartaglia, G.G.; Lehner, B. The mutational landscape of a prion-like domain. Nat. Commun. 2019, 10, 4162. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Lee, S.; Mallard, W.; Clement, K.; Tagliazucchi, G.M.; Lim, H.; Choi, I.Y.; Ferrari, F.; Tsankov, A.M.; Pop, R.; et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat. Biotechnol. 2015, 33, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Churko, J.M.; Burridge, P.W.; Wu, J.C. Generation of human iPSCs from human peripheral blood mononuclear cells using non-integrative Sendai virus in chemically defined conditions. Methods Mol. Biol. 2013, 1036, 81–88. [Google Scholar] [PubMed]
- Seminary, E.R.; Sison, S.L.; Ebert, A.D. Modeling Protein Aggregation and the Heat Shock Response in ALS iPSC-Derived Motor Neurons. Front. Neurosci. 2018, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, A.D.; Yu, J.; Rose, F.F.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Schwab, A.J.; Ebert, A.D. Neurite Aggregation and Calcium Dysfunction in iPSC-Derived Sensory Neurons with Parkinson’s Disease-Related LRRK2 G2019S Mutation. Stem Cell Rep. 2015, 5, 1039–1052. [Google Scholar] [CrossRef] [Green Version]
- Hosoyama, T.; McGivern, J.V.; Van Dyke, J.M.; Ebert, A.D.; Suzuki, M. Derivation of myogenic progenitors directly from human pluripotent stem cells using a sphere-based culture. Stem Cells Transl. Med. 2014, 3, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Sareen, D.; O’Rourke, J.G.; Meera, P.; Muhammad, A.K.; Grant, S.; Simpkinson, M.; Bell, S.; Carmona, S.; Ornelas, L.; Sahabian, A.; et al. Targeting RNA Foci in iPSC-Derived Motor Neurons from ALS Patients with a C9orf72 Repeat Expansion. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maury, Y.; Côme, J.; Piskorowski, R.A.; Salah-Mohellibi, N.; Chevaleyre, V.; Peschanski, M.; Martinat, C.; Nedelec, S. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 2015, 33, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Lieu, P.T.; Fontes, A.; Vemuri, M.C.; MacArthur, C.C. Generation of induced pluripotent stem cells with CytoTune, a non-integrating Sendai virus. Methods Mol. Biol. 2013, 997, 45–56. [Google Scholar] [PubMed]
- Xiao, Z.; Meng, Q.; Tsai, J.C.; Yuan, H.; Xu, N.; Li, Y. A novel optineurin genetic mutation associated with open-angle glaucoma in a Chinese family. Mol. Vis. 2009, 15, 1649–1654. [Google Scholar]
- Chu, T.T.; Liu, Y. An integrated genomic analysis of gene-function correlation on schizophrenia susceptibility genes. J. Hum. Genet. 2010, 55, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.G.; Kim, J.H.; Park, C.S.; Kim, B.J.; Kim, J.W.; Choi, I.G.; Hwang, J.; Shin, H.D.; Woo, S.I. Gender-Specific Associations between CHGB Genetic Variants and Schizophrenia in a Korean Population. Yonsei Med. J. 2017, 58, 619–625. [Google Scholar] [CrossRef]
- Blauw, H.M.; van Rheenen, W.; Koppers, M.; Van Damme, P.; Waibel, S.; Lemmens, R.; van Vught, P.W.; Meyer, T.; Schulte, C.; Gasser, T.; et al. NIPA1 polyalanine repeat expansions are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 2497–2502. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Hyeon, S.J.; Im, H.; Ryu, H.; Kim, Y.; Ryu, H. Astrocytes and Microglia as Non-cell Autonomous Players in the Pathogenesis of ALS. Exp. Neurobiol. 2016, 25, 233–240. [Google Scholar] [CrossRef]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.H.; Hung, S.T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef]
- Che, M.X.; Jiang, L.L.; Li, H.Y.; Jiang, Y.J.; Hu, H.Y. TDP-35 sequesters TDP-43 into cytoplasmic inclusions through binding with RNA. FEBS Lett. 2015, 589, 1920–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.J.; Xu, Y.F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef] [Green Version]
- Xiao, S.; Sanelli, T.; Chiang, H.; Sun, Y.; Chakrabartty, A.; Keith, J.; Rogaeva, E.; Zinman, L.; Robertson, J. Low molecular weight species of TDP-43 generated by abnormal splicing form inclusions in amyotrophic lateral sclerosis and result in motor neuron death. Acta Neuropathol. 2015, 130, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, M.X.; Jiang, Y.J.; Xie, Y.Y.; Jiang, L.L.; Hu, H.Y. Aggregation of the 35-kDa fragment of TDP-43 causes formation of cytoplasmic inclusions and alteration of RNA processing. FASEB J. 2011, 25, 2344–2353. [Google Scholar] [CrossRef]
- Burkhardt, M.F.; Martinez, F.J.; Wright, S.; Ramos, C.; Volfson, D.; Mason, M.; Garnes, J.; Dang, V.; Lievers, J.; Shoukat-Mumtaz, U.; et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci. 2013, 56, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, C.J.; Dariolli, R.; Jorge, F.M.D.H.; Monteiro, M.R.; Maximino, J.R.; Martins, R.S.; Strauss, B.E.; Krieger, J.E.; Callegaro, D.; Chadi, G. Gene expression profiling for human iPS-derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration. Front. Cell. Neurosci. 2015, 9, 289. [Google Scholar] [CrossRef] [Green Version]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef]
- Sun, X.; Song, J.; Huang, H.; Chen, H.; Qian, K. Modeling hallmark pathology using motor neurons derived from the family and sporadic amyotrophic lateral sclerosis patient-specific iPS cells. Stem Cell Res. Ther. 2018, 9, 315. [Google Scholar] [CrossRef] [Green Version]
- Logan, S.; Arzua, T.; Canfield, S.G.; Seminary, E.R.; Sison, S.L.; Ebert, A.D.; Bai, X. Studying Human Neurological Disorders Using Induced Pluripotent Stem Cells: From 2D Monolayer to 3D Organoid and Blood Brain Barrier Models. Compr. Physiol. 2019, 9, 565–611. [Google Scholar]
- Apostolou, E.; Hochedlinger, K. Chromatin dynamics during cellular reprogramming. Nature 2013, 502, 462–471. [Google Scholar] [CrossRef] [Green Version]
- Brennand, K.; Savas, J.N.; Kim, Y.; Tran, N.; Simone, A.; Hashimoto-Torii, K.; Beaumont, K.G.; Kim, H.J.; Topol, A.; Ladran, I.; et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry 2015, 20, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaguri, H.; Chew, J.; Xu, Y.F.; Gendron, T.F.; Garrett, A.; Lee, C.W.; Jansen-West, K.; Bauer, P.O.; Perkerson, E.A.; Tong, J.; et al. The extreme N-terminus of TDP-43 mediates the cytoplasmic aggregation of TDP-43 and associated toxicity in vivo. Brain Res. 2016, 1647, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillee, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kia, A.; McAvoy, K.; Krishnamurthy, K.; Trotti, D.; Pasinelli, P. Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia 2018, 66, 1016–1033. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.; Santos, C.; Gomes, C.; Fernandes, A.; Correia, A.M.; Sebastião, A.M.; Vaz, A.R.; Brites, D. Downregulated Glia Interplay and Increased miRNA-155 as Promising Markers to Track ALS at an Early Stage. Mol. Neurobiol. 2018, 55, 4207–4224. [Google Scholar] [CrossRef]
- Tripathi, P.; Rodriguez-Muela, N.; Klim, J.R.; de Boer, A.S.; Agrawal, S.; Sandoe, J.; Lopes, C.S.; Ogliari, K.S.; Williams, L.A.; Shear, M.; et al. Reactive Astrocytes Promote ALS-like Degeneration and Intracellular Protein Aggregation in Human Motor Neurons by Disrupting Autophagy through TGF-beta1. Stem Cell Rep. 2017, 9, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Pardo, A.C.; Wong, V.; Benson, L.M.; Dykes, M.; Tanaka, K.; Rothstein, J.D.; Maragakis, N.J. Loss of the astrocyte glutamate transporter GLT1 modifies disease in SOD1(G93A) mice. Exp. Neurol. 2006, 201, 120–130. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company | Catalog Number |
|---|---|---|
| Goat α-ChAT | Millipore | AB144P |
| Rabbit α-LC3 | Cell Signaling | 3868 |
| Rabbit α-Optineurin | abcam | ab151240 |
| Mouse α-p62 | BD Biosciences | 610832 |
| Rabbit α-SOD1 | abcam | ab13498 |
| Rabbit α-TDP-43 | abcam | ab109535 |
| Rabbit α-Tuj1 | Covance | MRB-435P |
| Mouse Islet1 | DSHB | 40.2D6 |
| Gene | Mutation | Genotype (Affected/ Unaffected) | Variant Type | Clinical Significance | gnomAD Frequency | HGMD Phenotype |
|---|---|---|---|---|---|---|
| CHGB | c.1058C>G p.Ala353Gly | Heterozygous/ Heterozygous | Missense | Variant of Unknown Significance | 0.451 | Schizophrenia |
| CHGB | c.1250G>A p.Arg417His | Heterozygous/ Heterozygous | Missense | Variant of Unknown Significance | 0.277 | Schizophrenia |
| OPTN | c.964A>G p.Lys322Glut | Homozygous/ Homozygous | Missense | Pathogenic | 0.997 | Open Angle Glaucoma |
| SIGMAR1 | c.*31A>G | Homozygous/ Homozygous | 3′ UTR variant | Variant of Unknown Significance | 0.995 | Amyotrophic lateral sclerosis |
| NIPA1 | c.42_47dupGGCGGC p.Ala15_Ala16dup | Heterozygous/ Heterozygous | Disruptive inframe insertion | Variant of Unknown Significance | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seminary, E.R.; Santarriaga, S.; Wheeler, L.; Mejaki, M.; Abrudan, J.; Demos, W.; Zimmermann, M.T.; Urrutia, R.A.; Fee, D.; Barkhaus, P.E.; et al. Motor Neuron Generation from iPSCs from Identical Twins Discordant for Amyotrophic Lateral Sclerosis. Cells 2020, 9, 571. https://doi.org/10.3390/cells9030571
Seminary ER, Santarriaga S, Wheeler L, Mejaki M, Abrudan J, Demos W, Zimmermann MT, Urrutia RA, Fee D, Barkhaus PE, et al. Motor Neuron Generation from iPSCs from Identical Twins Discordant for Amyotrophic Lateral Sclerosis. Cells. 2020; 9(3):571. https://doi.org/10.3390/cells9030571
Chicago/Turabian StyleSeminary, Emily R., Stephanie Santarriaga, Lynn Wheeler, Marie Mejaki, Jenica Abrudan, Wendy Demos, Michael T. Zimmermann, Raul A. Urrutia, Dominic Fee, Paul E. Barkhaus, and et al. 2020. "Motor Neuron Generation from iPSCs from Identical Twins Discordant for Amyotrophic Lateral Sclerosis" Cells 9, no. 3: 571. https://doi.org/10.3390/cells9030571