Alterations in Glucose Metabolism During the Transition to Heart Failure: The Contribution of UCP-2

 ,
,
Abstract
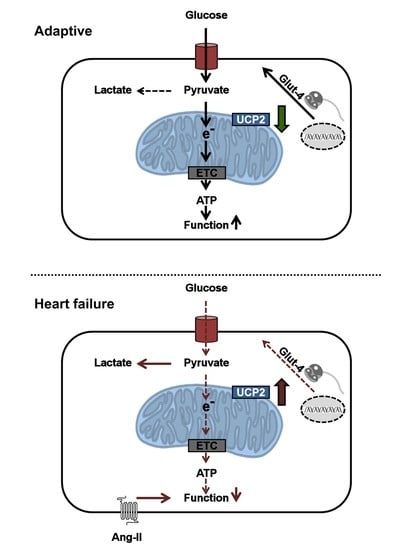
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Ethical Concerns
2.2. Isolation and Cultivation of Adult Rat Ventricular Cardiomyocytes
2.3. Isolation of Mitochondria and Measurement of ROS and Respiration
2.4. Glucose Uptake
2.5. Intracellular Lactate Levels
2.6. Cell Viability Assay
2.7. RNA Isolation and Real-Time PCR
2.8. Western Blots
2.9. Assessment of LV Hypertrophy and Function
2.10. Statistics
3. Results
3.1. Effect of UCP-2 Expression and Activity on Load-Free Cell Shortening of Adult Rat Ventricular Cardiomyocytes
3.2. Effects of UCP-2 Expression and Activity on Mitochondrial Function, Metabolism, and Viability
3.3. Regulation of UCP-2 and Glut-4 During Chronic Hypertension
3.4. Effect of Spironolactone on the Expression of UCP-2 and Glut-4 in Pressure-Overloaded Hearts
3.5. UCP-2 and Glut-4 in Human End-Stage Heart Failure
4. Discussion
5. Study Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ang-II | angiotensin II |
| ANP | atrial natriuretic peptide |
| EF | ejection fraction |
| FCS | fetal calf serum |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| Glut-4 | glucose transporter type 4 |
| HepG2 | hepatocelluar carcinoma |
| Hprt | hypoxanthine phosphoribosyl transferase |
| hPSC | human pluripotent stem cells |
| hUCP2 | human UCP-2 protein |
| LVW/TL | left ventricular weight normalized to tibia length |
| MRA | mineralocorticoid receptor antagonist |
| PBS | phosphate buffered saline |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| RAAS | renin-angiotensin-aldosterone-system |
| ROS | reactive oxygen species |
| SD | standard deviation |
| SE | standard error |
| scRNA | scrambled RNA (control) |
| SHR | spontaneously hypertensive rats |
| siRNA | small interfering RNA |
| UCP-2 | uncoupling protein 2 |
References
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar]
- Azzu, V.; Jastroch, M.; Divakaruni, A.S.; Brand, M.D. The regulation and turnover of mitochondrial uncoupling proteins. Biochim. Biophys. Acta 2010, 1797, 785–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteves, T.C.; Brand, M.D. The reactions catalysed by the mitochondrial uncoupling proteins UCP2 and UCP3. Biochim. Biophys. Acta 2005, 1709, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esfandiary, A.; Kutsche, H.; Schreckenberg, R.; Weber, M.; Pak, O.; Kojonazarov, B.; Sydykov, A.; Hirschhäuser, C.; Wolf, A.; Haag, D.; et al. Protection against pressure overload-induced right heart failure by uncoupling protein 2 silencing. Cardiovasc. Res. 2019, 115, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Mattiasson, G.; Sullivan, P.G. The emerging functions of UCP2 in health, disease, and therapeutics. Antioxid. Redox Signal. 2006, 8, 1–38. [Google Scholar] [CrossRef]
- Noma, T. Possible role of uncoupling protein in regulation of myocardial energy metabolism in aortic regurgitation model rats. FASEB J. 2001, 7, 1206–1208. [Google Scholar] [CrossRef]
- Murray, A.J.; Cole, M.A.; Lygate, C.A.; Carr, C.A.; Stuckey, D.J.; Little, S.E.; Neubauer, S.; Clarke, K. Increased mitochondrial uncoupling proteins, respiratory uncoupling and decreased efficiency in the chronically infarcted rat heart. J. Mol. Cell. Cardiol. 2008, 44, 694–700. [Google Scholar] [CrossRef]
- Ji, X.-B.; Li, X.-R.; Hao-Ding; Sun, Q.; Zhou, Y.; Wen, P.; Dai, C.-S.; Yang, J.-W. Inhibition of Uncoupling Protein 2 Attenuates Cardiac Hypertrophy Induced by Transverse Aortic Constriction in Mice. Cell. Physiol. Biochem. 2015, 36, 1688–1698. [Google Scholar] [CrossRef]
- Rohrbach, S.; Niemann, B.; Silber, R.-E.; Holtz, J. Neuregulin receptors erbB2 and erbB4 in failing human myocardium—depressed expression and attenuated activation. Basic Res. Cardiol. 2005, 100, 240–249. [Google Scholar] [CrossRef]
- Nippert, F.; Schreckenberg, R.; Schlüter, K.-D. Isolation and Cultivation of Adult Rat Cardiomyocytes. J. Vis. Exp. 2017, 128, e56634. [Google Scholar] [CrossRef]
- Bøtker, H.E.; Hausenloy, D.; Andreadou, I.; Antonucci, S.; Boengler, K.; Davidson, S.M.; Deshwal, S.; Devaux, Y.; Di Lisa, F.; Di Sante, M.; et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res. Cardiol. 2018, 113, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, M.; Lüttecke, D.; Schlüter, K.-D. Mechanism of the positive contractile effect of nitric oxide on rat ventricular cardiomyocytes with positive force/frequency relationship. Pfülgers Arch. Eur. J. Physiol. 2003, 447, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Holmuhamedov, E.L.; Jovanović, S.; Dzeja, P.P.; Jovanović, A.; Terzic, A. Mitochondrial ATP-sensitive K+ channels modulate cardiac mitochondrial function. Am. J. Physiol. 1998, 275, H1567–H1576. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Bulic, M.; Schreckenberg, R.; Schlüter, K.-D.; Schulz, R. The gap junction modifier ZP1609 decreases cardiomyocyte hypercontracture following ischaemia/reperfusion independent from mitochondrial connexin 43. Br. J. Pharmacol. 2017, 174, 2060–2073. [Google Scholar] [CrossRef]
- Nippert, F.; Schreckenberg, R.; Hess, A.; Weber, M.; Schlüter, K.-D. The Effects of Swiprosin-1 on the Formation of Pseudopodia-Like Structures and β-Adrenoceptor Coupling in Cultured Adult Rat Ventricular Cardiomyocytes. PLoS ONE 2016, 11, e0167655. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Rupprecht, A.; Sokolenko, E.A.; Beck, V.; Ninnemann, O.; Jaburek, M.; Trimbuch, T.; Klishin, S.S.; Jezek, P.; Skulachev, V.P.; Pohl, E.E. Role of the transmembrane potential in the membrane proton leak. Biophys. J. 2010, 98, 1503–1511. [Google Scholar] [CrossRef] [Green Version]
- Schreckenberg, R.; Horn, A.-M.; da Costa Rebelo, R.M.; Simsekyilmaz, S.; Niemann, B.; Li, L.; Rohrbach, S.; Schlüter, K.-D. Effects of 6-months’ Exercise on Cardiac Function, Structure and Metabolism in Female Hypertensive Rats–The Decisive Role of Lysyl Oxidase and Collagen III. Front. Physiol. 2017, 8, 1054. [Google Scholar] [CrossRef] [Green Version]
- Abu-Elheiga, L.; Oh, W.; Kordari, P.; Wakil, S.J. Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets. Proc. Natl. Acad. Sci. USA 2003, 100, 10207–10212. [Google Scholar] [CrossRef] [Green Version]
- Pecqueur, C.; Bui, T.; Gelly, C.; Hauchard, J.; Barbot, C.; Bouillaud, F.; Ricquier, D.; Miroux, B.; Thompson, C.B. Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J. 2008, 22, 9–18. [Google Scholar] [CrossRef]
- Vozza, A.; Parisi, G.; de Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, J.; Muthusamy, T.; Srinivasan, C.; Balasubramanian, K. Impact of excess aldosterone on glucose homeostasis in adult male rat. Clin. Chim. Acta 2009, 407, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Lastra, G.; Whaley-Connell, A.; Manrique, C.; Habibi, J.; Gutweiler, A.A.; Appesh, L.; Hayden, M.R.; Wei, Y.; Ferrario, C.; Sowers, J.R. Low-dose spironolactone reduces reactive oxygen species generation and improves insulin-stimulated glucose transport in skeletal muscle in the TG(mRen2)27 rat. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E110–E116. [Google Scholar] [CrossRef] [PubMed]
- Raheja, P.; Price, A.; Wang, Z.; Arbique, D.; Adams-Huet, B.; Auchus, R.J.; Vongpatanasin, W. Spironolactone prevents chlorthalidone-induced sympathetic activation and insulin resistance in hypertensive patients. Hypertension 2012, 60, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Alán, L.; Smolková, K.; Kronusová, E.; Santorová, J.; Jezek, P. Absolute levels of transcripts for mitochondrial uncoupling proteins UCP2, UCP3, UCP4, and UCP5 show different patterns in rat and mice tissues. J. Bioenerg. Biomembr. 2009, 41, 71–78. [Google Scholar] [CrossRef]
- Roshon, M.J.; Kline, J.A.; Thornton, L.R.; Watts, J.A. Cardiac UCP2 expression and myocardial oxidative metabolism during acute septic shock in the rat. Shock 2003, 19, 570–576. [Google Scholar] [CrossRef]
- Ruiz-Ramírez, A.; López-Acosta, O.; Barrios-Maya, M.A.; El-Hafidi, M. Cell Death and Heart Failure in Obesity: Role of Uncoupling Proteins. Oxid. Med. Cell. Longev. 2016, 2016, 9340654. [Google Scholar] [CrossRef] [Green Version]
- Teshima, Y.; Akao, M.; Jones, S.P.; Marbán, E. Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ. Res. 2003, 93, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Harper, M.-E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Y.; Guo, J.; Jin, K.; Li, J.; Guo, X.; Scott, G.I.; Zheng, Q.; Ren, J. Inhibition of protein kinase C βII isoform rescues glucose toxicity-induced cardiomyocyte contractile dysfunction: Role of mitochondria. Life Sci. 2013, 93, 116–124. [Google Scholar] [CrossRef]
- Kukat, A.; Dogan, S.A.; Edgar, D.; Mourier, A.; Jacoby, C.; Maiti, P.; Mauer, J.; Becker, C.; Senft, K.; Wibom, R.; et al. Loss of UCP2 attenuates mitochondrial dysfunction without altering ROS production and uncoupling activity. PLoS Genet. 2014, 10, e1004385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Xu, X.; Nie, L.; Xiao, T.; Guan, X.; He, T.; Yu, Y.; Liu, L.; Huang, Y.; Zhang, J.; et al. Indoxyl sulfate induces oxidative stress and hypertrophy in cardiomyocytes by inhibiting the AMPK/UCP2 signaling pathway. Toxicol. Lett. 2015, 234, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, K.A.; Willemsen, P.H.; van der Vusse, G.J.; van Bilsen, M. Effects of fatty acids on uncoupling protein-2 expression in the rat heart. FASEB J. 2000, 14, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vettor, R.; Fabris, R.; Serra, R.; Lombardi, A.M.; Tonello, C.; Granzotto, M.; Marzolo, M.O.; Carruba, M.O.; Ricquier, D.; Federspil, G.; et al. Changes in FAT/CD36, UCP2, UCP3 and GLUT4 gene expression during lipid infusion in rat skeletal and heart muscle. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 838–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Suwa, A.; Shimokawa, T. Glucose catabolic gene mRNA levels in skeletal muscle exhibit non-coordinate expression in hyperglycemic mice. Horm. Metab. Res. 2004, 36, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Fabris, R.; Nisoli, E.; Lombardi, A.M.; Tonello, C.; Serra, R.; Granzotto, M.; Cusin, I.; Rohner-Jeanrenaud, F.; Federspil, G.; Carruba, M.O.; et al. Preferential channeling of energy fuels toward fat rather than muscle during high free fatty acid availability in rats. Diabetes 2001, 50, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Cha, B.-Y.; Choi, S.-S.; Harada, Y.; Choi, B.-K.; Yonezawa, T.; Teruya, T.; Nagai, K.; Woo, J.-T. Fargesin improves lipid and glucose metabolism in 3T3-L1 adipocytes and high-fat diet-induced obese mice. Biofactors 2012, 38, 300–308. [Google Scholar] [CrossRef]
- McAllan, L.; Skuse, P.; Cotter, P.D.; O’Connor, P.; Cryan, J.F.; Ross, R.P.; Fitzgerald, G.; Roche, H.M.; Nilaweera, K.N. Protein quality and the protein to carbohydrate ratio within a high fat diet influences energy balance and the gut microbiota in C57BL/6J mice. PLoS ONE 2014, 9, e88904. [Google Scholar] [CrossRef] [Green Version]
- Samadder, A.; Das, J.; Das, S.; De, A.; Saha, S.K.; Bhattacharyya, S.S.; Khuda-Bukhsh, A.R. Poly(lactic-co-glycolic) acid loaded nano-insulin has greater potentials of combating arsenic induced hyperglycemia in mice: Some novel findings. Toxicol. Appl. Pharmacol. 2013, 267, 57–73. [Google Scholar] [CrossRef]
- Samadder, A.; Das, S.; Das, J.; Khuda-Bukhsh, A.R. Relative efficacies of insulin and poly (lactic-co-glycolic) acid encapsulated nano-insulin in modulating certain significant biomarkers in arsenic intoxicated L6 cells. Colloids Surf. B Biointerfaces 2013, 109, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.J.; Anderson, R.E.; Watson, G.C.; Radda, G.K.; Clarke, K. Uncoupling proteins in human heart. Lancet 2004, 364, 1786–1788. [Google Scholar] [CrossRef]
- Neves, F.A.; Cortez, E.; Bernardo, A.F.; Mattos, A.B.M.; Vieira, A.K.; Malafaia, T.d.O.; Thole, A.A.; Rodrigues-Cunha, A.C.d.S.; Garcia-Souza, E.P.; Sichieri, R.; et al. Heart energy metabolism impairment in Western-diet induced obese mice. J. Nutr. Biochem. 2014, 25, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef] [Green Version]
- Bodyak, N.; Rigor, D.L.; Chen, Y.-S.; Han, Y.; Bisping, E.; Pu, W.T.; Kang, P.M. Uncoupling protein 2 modulates cell viability in adult rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H829–H835. [Google Scholar] [CrossRef]
- Fukunaga, Y.; Itoh, H.; Hosoda, K.; Doi, K.; Matsuda, J.; Son, C.; Yamashita, J.; Chun, T.H.; Tanaka, T.; Inoue, M.; et al. Altered gene expression of uncoupling protein-2 and -3 in stroke-prone spontaneously hypertensive rats. J. Hypertens. 2000, 18, 1233–1238. [Google Scholar] [CrossRef]
- Li, P.; Guo, X.; Lei, P.; Shi, S.; Luo, S.; Cheng, X. PI3K/Akt/uncoupling protein 2 signaling pathway may be involved in cell senescence and apoptosis induced by angiotensin II in human vascular endothelial cells. Mol. Biol. Rep. 2014, 41, 6931–6937. [Google Scholar] [CrossRef]
- Mufti, S.; Wenzel, S.; Euler, G.; Piper, H.M.; Schlüter, K.-D. Angiotensin II-dependent loss of cardiac function: Mechanisms and pharmacological targets attenuating this effect. J. Cell. Physiol. 2008, 217, 242–249. [Google Scholar] [CrossRef]
- Deng, M.; Wang, D.; He, S.; Xu, R.; Xie, Y. SIRT1 confers protection against ischemia/reperfusion injury in cardiomyocytes via regulation of uncoupling protein 2 expression. Mol. Med. Rep. 2017, 16, 7098–7104. [Google Scholar] [CrossRef]
- Razeghi, P.; Young, M.E.; Ying, J.; Depre, C.; Uray, I.P.; Kolesar, J.; Shipley, G.L.; Moravec, C.S.; Davies, P.J.A.; Frazier, O.H.; et al. Downregulation of metabolic gene expression in failing human heart before and after mechanical unloading. Cardiology 2002, 97, 203–209. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutsche, H.S.; Schreckenberg, R.; Weber, M.; Hirschhäuser, C.; Rohrbach, S.; Li, L.; Niemann, B.; Schulz, R.; Schlüter, K.-D. Alterations in Glucose Metabolism During the Transition to Heart Failure: The Contribution of UCP-2. Cells 2020, 9, 552. https://doi.org/10.3390/cells9030552
Kutsche HS, Schreckenberg R, Weber M, Hirschhäuser C, Rohrbach S, Li L, Niemann B, Schulz R, Schlüter K-D. Alterations in Glucose Metabolism During the Transition to Heart Failure: The Contribution of UCP-2. Cells. 2020; 9(3):552. https://doi.org/10.3390/cells9030552
Chicago/Turabian StyleKutsche, Hanna Sarah, Rolf Schreckenberg, Martin Weber, Christine Hirschhäuser, Susanne Rohrbach, Ling Li, Bernd Niemann, Rainer Schulz, and Klaus-Dieter Schlüter. 2020. "Alterations in Glucose Metabolism During the Transition to Heart Failure: The Contribution of UCP-2" Cells 9, no. 3: 552. https://doi.org/10.3390/cells9030552